Department of Computational Medicine and Bioinformatics, University of Michigan, 100 Washtenaw Avenue, Ann Arbor, MI, 48109, USA.
Department of Internal Medicine, University of Michigan, 100 Washtenaw Avenue, Ann Arbor, MI, 48109, USA.
BMC Biol. 2019 Dec 23;17(1):107. doi: 10.1186/s12915-019-0730-9.
The classic central dogma in biology is the information flow from DNA to mRNA to protein, yet complicated regulatory mechanisms underlying protein translation often lead to weak correlations between mRNA and protein abundances. This is particularly the case in cancer samples and when evaluating the same gene across multiple samples.
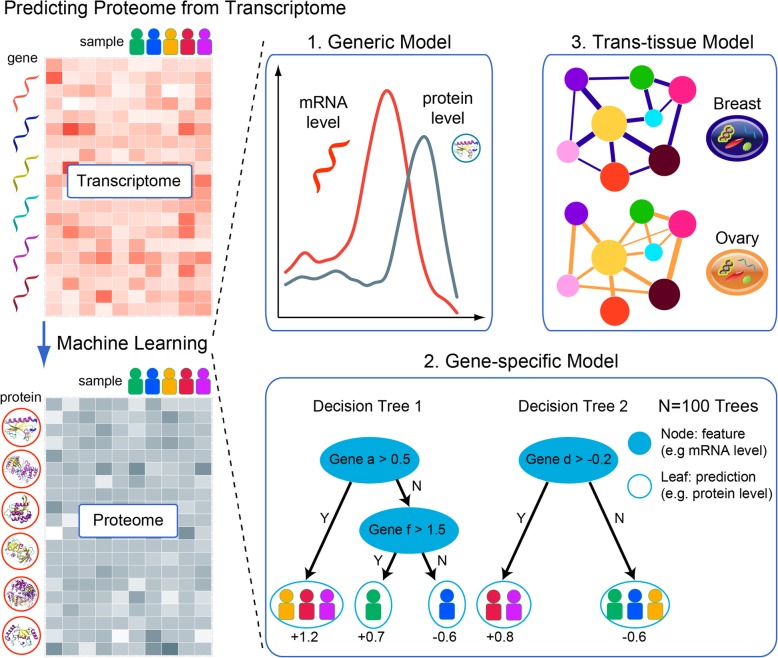
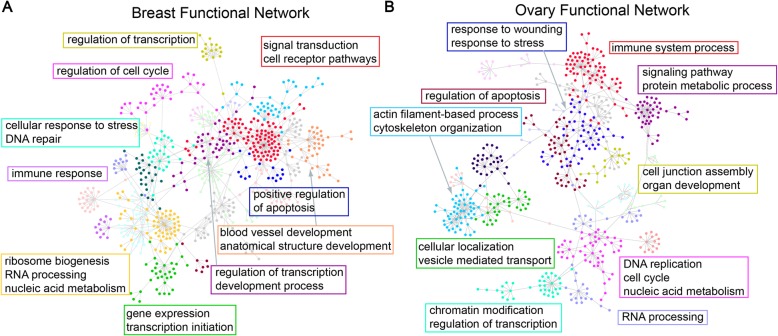
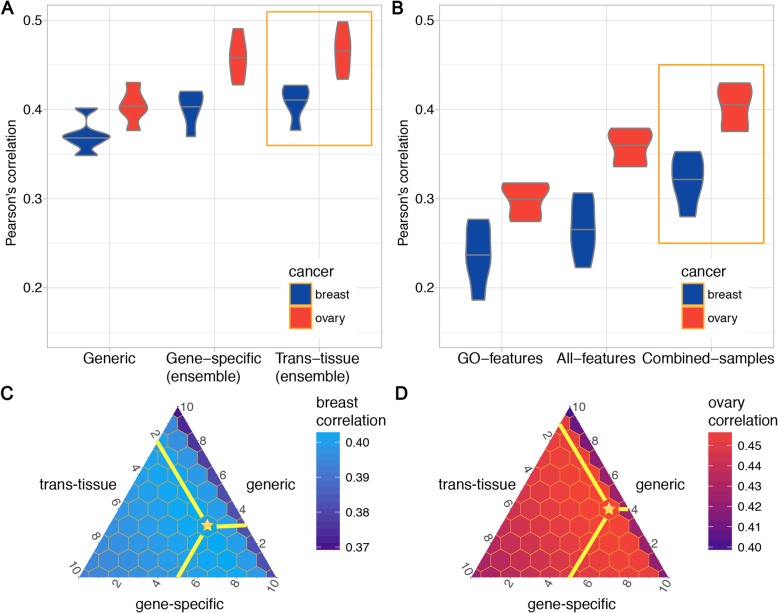
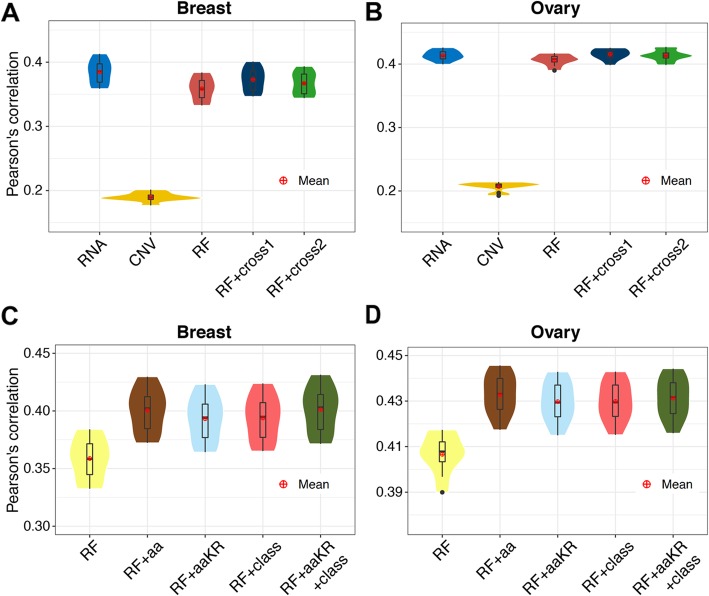
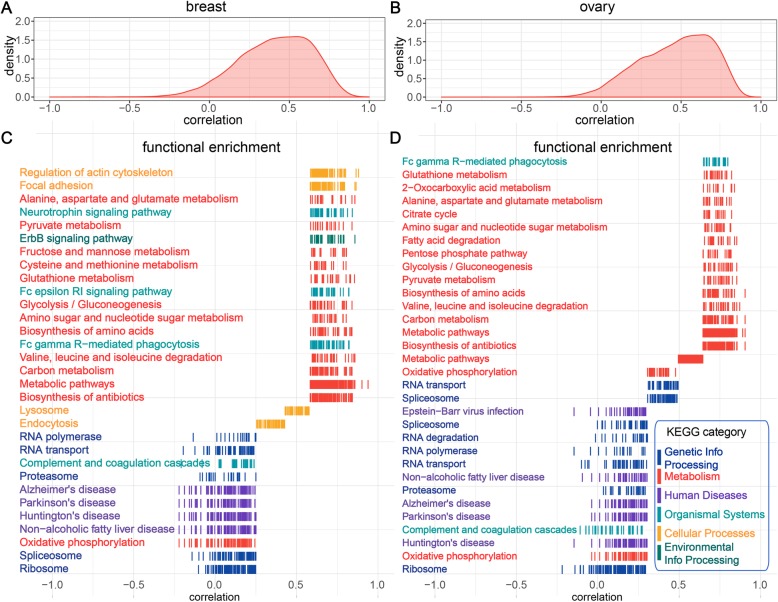
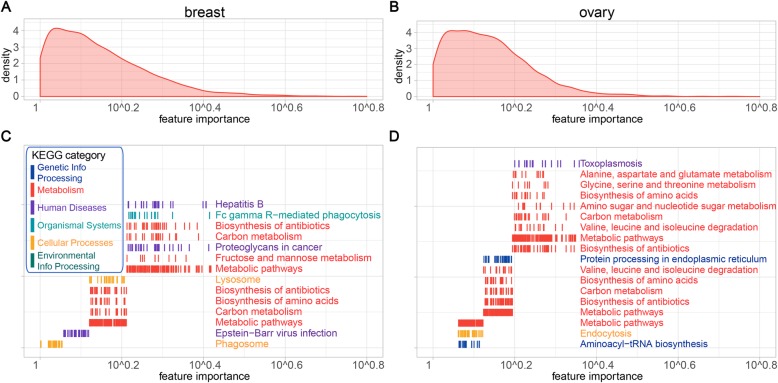
Here, we report a method for predicting proteome from transcriptome, using a training dataset provided by NCI-CPTAC and TCGA, consisting of transcriptome and proteome data from 77 breast and 105 ovarian cancer samples. First, we establish a generic model capturing the correlation between mRNA and protein abundance of a single gene. Second, we build a gene-specific model capturing the interdependencies among multiple genes in a regulatory network. Third, we create a cross-tissue model by joint learning the information of shared regulatory networks and pathways across cancer tissues. Our method ranked first in the NCI-CPTAC DREAM Proteogenomics Challenge, and the predictive performance is close to the accuracy of experimental replicates. Key functional pathways and network modules controlling the proteomic abundance in cancers were revealed, in particular metabolism-related genes.
We present a method to predict proteome from transcriptome, leveraging data from different cancer tissues to build a trans-tissue model, and suggest how to integrate information from multiple cancers to provide a foundation for further research.
生物学中的经典中心法则是指信息从 DNA 流向 mRNA 再流向蛋白质,然而,蛋白质翻译背后复杂的调控机制常常导致 mRNA 和蛋白质丰度之间的相关性较弱。在癌症样本中以及在评估多个样本中的相同基因时,这种情况尤其明显。
在这里,我们报告了一种使用 NCI-CPTAC 和 TCGA 提供的训练数据集从转录组预测蛋白质组的方法,该数据集由 77 个乳腺癌和 105 个卵巢癌样本的转录组和蛋白质组数据组成。首先,我们建立了一个通用模型,该模型捕获了单个基因的 mRNA 和蛋白质丰度之间的相关性。其次,我们构建了一个基因特异性模型,该模型捕获了调控网络中多个基因之间的相互依赖关系。第三,我们通过联合学习癌症组织中共享调控网络和途径的信息来创建跨组织模型。我们的方法在 NCI-CPTAC DREAM Proteogenomics 挑战赛中排名第一,其预测性能接近实验复制品的准确性。揭示了控制癌症中蛋白质组丰度的关键功能途径和网络模块,特别是与代谢相关的基因。
我们提出了一种从转录组预测蛋白质组的方法,利用来自不同癌症组织的数据构建跨组织模型,并提出如何整合来自多个癌症的信息,为进一步研究提供基础。