Department of Medicine/Cardiology, University of Colorado School of Medicine, Aurora, Colorado, United States of America.
Consortium for Fibrosis Research and Translation, University of Colorado School of Medicine, Aurora, Colorado, United States of America.
PLoS Comput Biol. 2022 Nov 10;18(11):e1010702. doi: 10.1371/journal.pcbi.1010702. eCollection 2022 Nov.
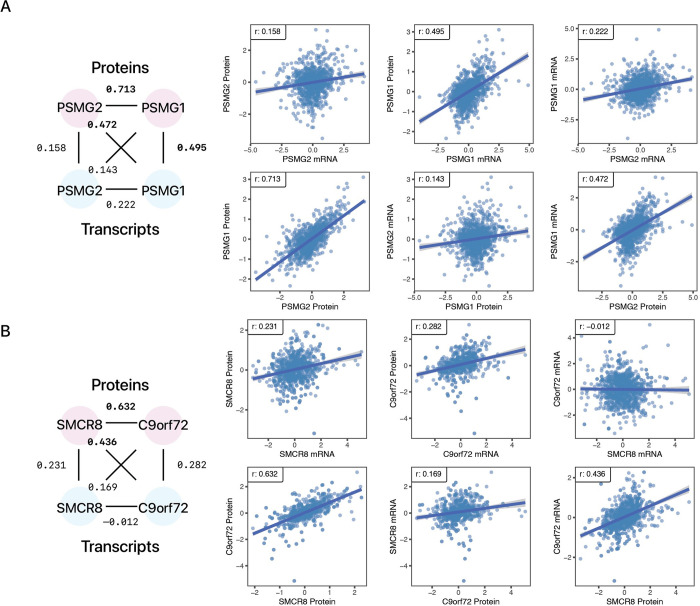
Protein and mRNA levels correlate only moderately. The availability of proteogenomics data sets with protein and transcript measurements from matching samples is providing new opportunities to assess the degree to which protein levels in a system can be predicted from mRNA information. Here we examined the contributions of input features in protein abundance prediction models. Using large proteogenomics data from 8 cancer types within the Clinical Proteomic Tumor Analysis Consortium (CPTAC) data set, we trained models to predict the abundance of over 13,000 proteins using matching transcriptome data from up to 958 tumor or normal adjacent tissue samples each, and compared predictive performances across algorithms, data set sizes, and input features. Over one-third of proteins (4,648) showed relatively poor predictability (elastic net r ≤ 0.3) from their cognate transcripts. Moreover, we found widespread occurrences where the abundance of a protein is considerably less well explained by its own cognate transcript level than that of one or more trans locus transcripts. The incorporation of additional trans-locus transcript abundance data as input features increasingly improved the ability to predict sample protein abundance. Transcripts that contribute to non-cognate protein abundance primarily involve those encoding known or predicted interaction partners of the protein of interest, including not only large multi-protein complexes as previously shown, but also small stable complexes in the proteome with only one or few stable interacting partners. Network analysis further shows a complex proteome-wide interdependency of protein abundance on the transcript levels of multiple interacting partners. The predictive model analysis here therefore supports that protein-protein interaction including in small protein complexes exert post-transcriptional influence on proteome compositions more broadly than previously recognized. Moreover, the results suggest mRNA and protein co-expression analysis may have utility for finding gene interactions and predicting expression changes in biological systems.
蛋白质和 mRNA 水平的相关性仅为中等。具有来自匹配样本的蛋白质和转录物测量的蛋白质基因组学数据集的可用性为评估系统中蛋白质水平从 mRNA 信息预测的程度提供了新的机会。在这里,我们研究了输入特征在蛋白质丰度预测模型中的贡献。使用来自临床蛋白质组肿瘤分析联盟(CPTAC)数据集中 8 种癌症类型的大型蛋白质基因组学数据,我们使用来自每个肿瘤或正常相邻组织样本的多达 958 个转录组数据训练了模型,以预测超过 13000 种蛋白质的丰度,并比较了算法,数据集大小和输入特征的预测性能。超过三分之一的蛋白质(4648 种)显示出与其同源转录物相对较差的可预测性(弹性网络 r ≤ 0.3)。此外,我们发现广泛存在一种情况,即蛋白质的丰度与其自身同源转录物水平相比,由一个或多个跨基因座转录物解释的程度要差得多。作为输入特征纳入额外的跨基因座转录物丰度数据可逐渐提高预测样品蛋白质丰度的能力。有助于非同源蛋白质丰度的转录物主要涉及那些编码感兴趣蛋白质的已知或预测的相互作用伙伴的转录物,包括不仅像以前那样包括大的多蛋白复合物,而且还包括蛋白质组中具有一个或几个稳定相互作用伙伴的小稳定复合物。网络分析进一步显示了蛋白质丰度与多个相互作用伙伴的转录水平之间的广泛的复杂的全蛋白质组相互依存关系。因此,这里的预测模型分析支持蛋白质 - 蛋白质相互作用,包括小蛋白质复合物,对蛋白质组组成的转录后影响比以前认识的更广泛。此外,这些结果表明,mRNA 和蛋白质共表达分析可能有助于发现基因相互作用并预测生物系统中的表达变化。