Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, Maryland.
Department of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland.
Ann Clin Transl Neurol. 2020 Feb;7(2):254-258. doi: 10.1002/acn3.50979. Epub 2020 Jan 10.
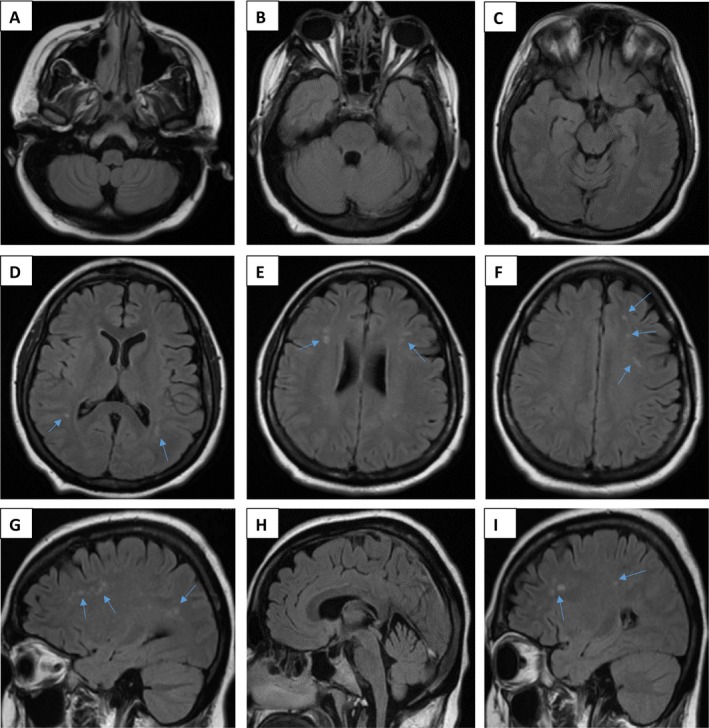
Aicardi-Goutières syndrome (AGS) is a rare and likely underdiagnosed genetic leukoencephalopathy, typically presenting in infancy with encephalopathy and characteristic neuroimaging features, with residual static neurological deficits. We describe a patient who, following an initial presentation at the age of 12 months in keeping with AGS, exhibited a highly atypical relapsing course of neurological symptoms in adulthood with essentially normal neuroimaging. Whole-exome sequencing confirmed a pathogenic RNASEH2B gene variant consistent with AGS. This case highlights the expanding phenotypes associated with AGS and the potential role of whole-exome sequencing in facilitating an increase in the rate of diagnosis.
Aicardi-Goutières 综合征(AGS)是一种罕见且可能诊断不足的遗传性脑白质病,通常在婴儿期表现为脑病和特征性神经影像学特征,伴有残留的静态神经功能缺损。我们描述了一位患者,其在 12 个月大时首次出现符合 AGS 的表现,随后在成年期表现出高度非典型的复发性神经症状,神经影像学基本正常。全外显子组测序证实存在 RNASEH2B 基因变异,与 AGS 一致。该病例强调了与 AGS 相关的表型不断扩大,以及全外显子组测序在提高诊断率方面的潜在作用。