Department of Rheumatology and Immunology, Shenzhen Children's Hospital, 7019 Yitian Road, Shenzhen, 518038, China.
Pediatr Rheumatol Online J. 2021 Jan 22;19(1):9. doi: 10.1186/s12969-021-00497-2.
Aicardi-Goutières (AGS) is a rare immune dysregulated disease due to mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, or IFIH1. Clinical features include basal ganglia calcifications, white matter abnormalities, and cerebral atrophy. Severe systemic inflammation and chronic kidney disease (CKD) are extremely rare in AGS. Herein, we report a patient presenting with systemic inflammation and CKD to broaden the clinical phenotype spectrum of the RNASEH2B defect.
All testing and molecular genetic analysis were performed after obtaining the informed consent of the parents. Demographic, clinical, and laboratory findings were abstracted from outpatient and inpatient encounters. Cerebral magnetic resonance imaging (MRI), computed tomography (CT) scans, and renal biopsy histopathology reports were reviewed and summarized. Whole exome sequencing (WES) was performed on peripheral blood cells. After exposure to cGAMP in vitro for 24 h, mRNA expression of 12 IFN-stimulated cytokine genes in PBMCs was assessed. Serum cytokine levels were detected by Milliplex.
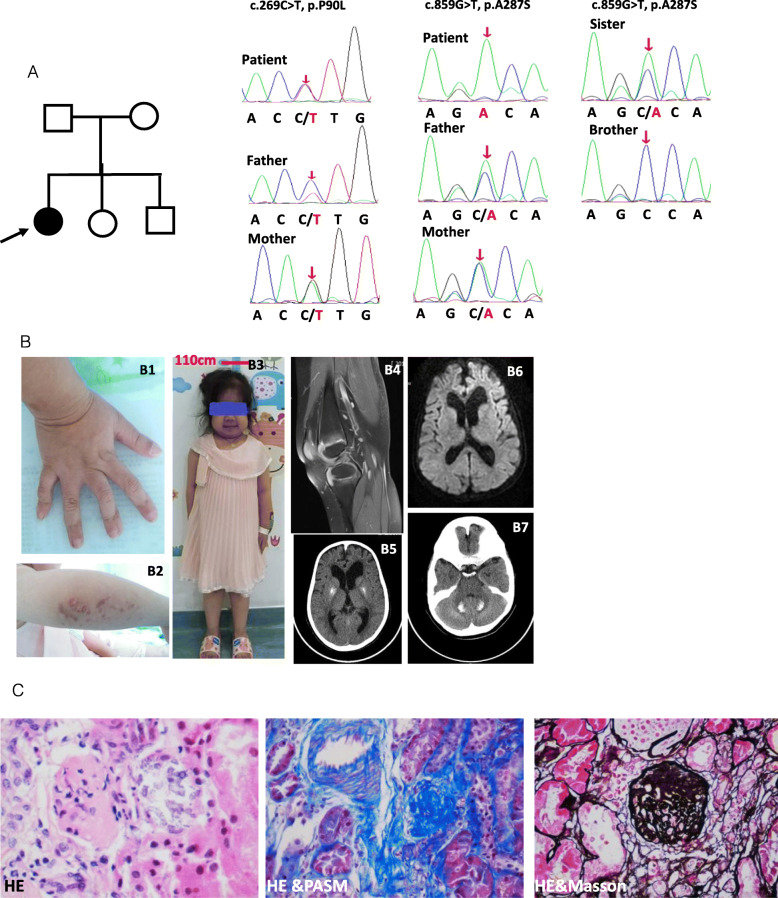
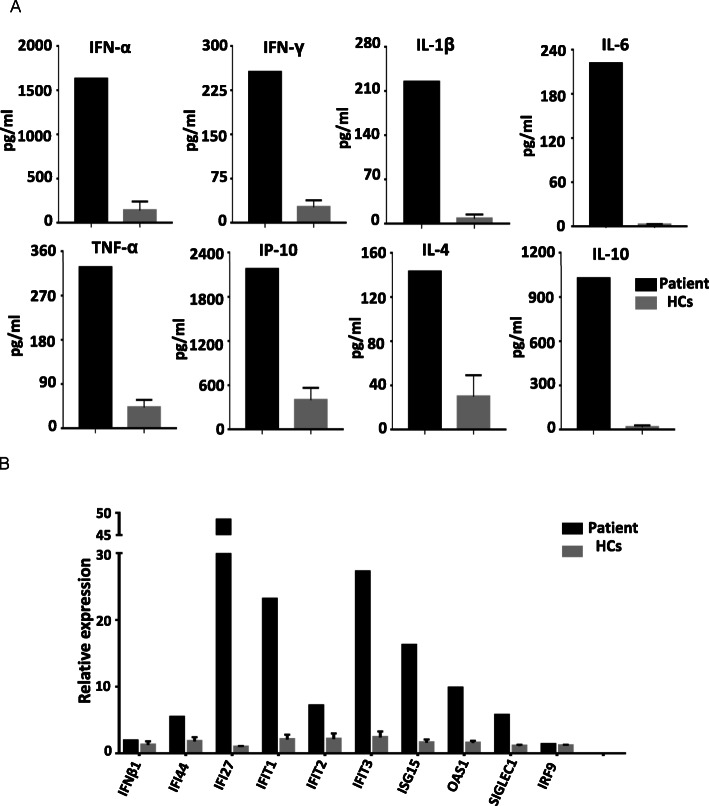
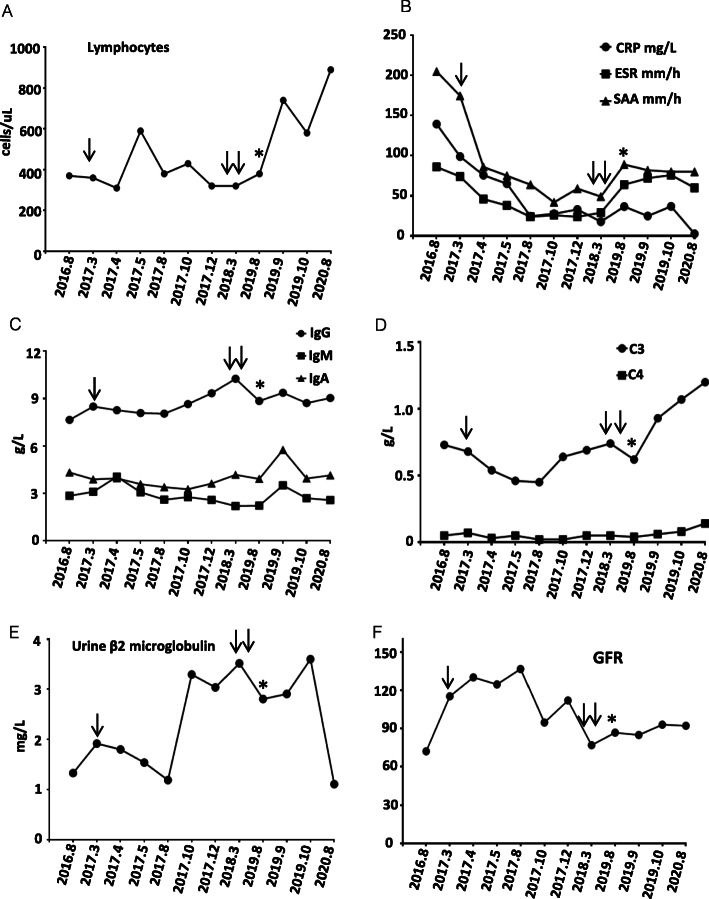
A 11-year-old girl presented with recurrent aseptic fever, arthritis, chilblains, failure to thrive, mild hearing loss, and neurological manifestations. Laboratory and immunologic findings demonstrated lymphopenia, low complement levels, positive autoantibodies, elevated levels of acute-phase reactants and inflammatory cytokines. Cerebral imaging showed cerebral atrophy, white matter abnormalities, and intracranial calcification. Renal biopsy showed glomerular sclerosis in 3 of 14 glomeruli, infiltration of lymphocytes and other mononuclear cells. WES revealed a homozygous and heterozygous mutations in RNASEH2B. Over-expression of IFN-stimulated cytokine genes was observed, including IFI44, IFI27, IFIT1, IFIT2, IFIT3, ISG15, OAS1, and SIGLEC1.
To date, only two cases with AGS have been reported to have renal disease. Here, we describe a patient with both homozygous and heterozygous variants in RNASEH2B, presenting with neurological manifestations, persistently systemic autoinflammation, and CKD. CKD has never been reported in patients with AGS due to the RNASEH2B defect.
Not applicable; this was a retrospective study.
Aicardi-Goutières(AGS)是一种罕见的免疫失调疾病,由 TREX1、RNASEH2A、RNASEH2B、RNASEH2C、SAMHD1、ADAR1 或 IFIH1 基因突变引起。临床特征包括基底节钙化、白质异常和脑萎缩。AGS 患者极罕见出现严重的全身炎症和慢性肾脏病(CKD)。本文报道了一例以全身炎症和 CKD 为表现的患者,以拓宽 RNASEH2B 缺陷的临床表型谱。
在获得父母知情同意后,进行所有检测和分子遗传学分析。从门诊和住院病历中提取人口统计学、临床和实验室检查结果。回顾和总结脑磁共振成像(MRI)、计算机断层扫描(CT)扫描和肾活检组织病理学报告。对外周血单个核细胞(PBMC)进行全外显子组测序(WES)。体外用 cGAMP 孵育 24 小时后,评估 PBMC 中 12 种 IFN 刺激细胞因子基因的 mRNA 表达。通过 Milliplex 检测血清细胞因子水平。
一名 11 岁女孩因反复无菌性发热、关节炎、冻疮、生长发育迟缓、轻度听力损失和神经系统表现就诊。实验室和免疫检查发现淋巴细胞减少、补体水平降低、自身抗体阳性、急性期反应物和炎症细胞因子水平升高。脑部影像学显示脑萎缩、白质异常和颅内钙化。肾活检显示 14 个肾小球中有 3 个肾小球硬化,淋巴细胞和其他单核细胞浸润。WES 显示 RNASEH2B 存在纯合和杂合突变。IFN 刺激细胞因子基因的过表达,包括 IFI44、IFI27、IFIT1、IFIT2、IFIT3、ISG15、OAS1 和 SIGLEC1。
迄今为止,仅有两例 AGS 患者报道有肾脏疾病。本研究描述了一例 RNASEH2B 既有纯合突变又有杂合突变的患者,表现为神经系统表现、持续性全身自身炎症和 CKD。AGS 患者由于 RNASEH2B 缺陷,CKD 从未被报道过。
不适用;这是一项回顾性研究。