Department of Medicinal Chemistry, Uppsala University, BMC, Box 574, 751 23, Uppsala, Sweden.
Medicinal Chemistry, Research and Early Development Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden.
J Comput Aided Mol Des. 2020 Mar;34(3):231-252. doi: 10.1007/s10822-020-00277-2. Epub 2020 Jan 21.
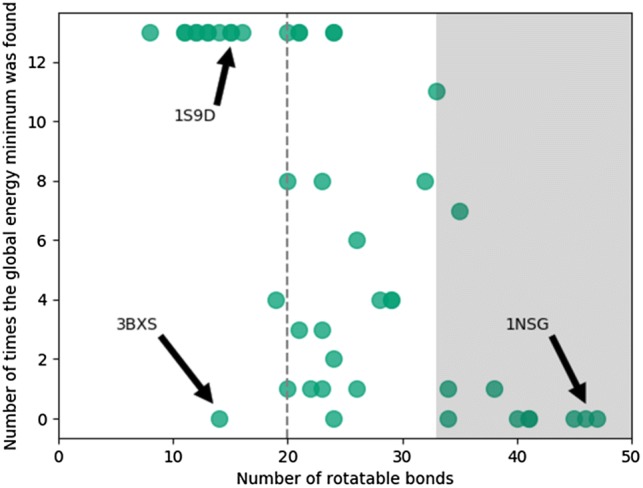
Macrocycles represent an important class of medicinally relevant small molecules due to their interesting biological properties. Therefore, a firm understanding of their conformational preferences is important for drug design. Given the importance of macrocycle-protein modelling in drug discovery, we envisaged that a systematic study of both classical and recent specialized methods would provide guidance for other practitioners within the field. In this study we compare the performance of the general, well established conformational analysis methods Monte Carlo Multiple Minimum (MCMM) and Mixed Torsional/Low-Mode sampling (MTLMOD) with two more recent and specialized macrocycle sampling techniques: MacroModel macrocycle Baseline Search (MD/LLMOD) and Prime macrocycle conformational sampling (PRIME-MCS). Using macrocycles extracted from 44 macrocycle-protein X-ray crystallography complexes, we evaluated each method based on their ability to (i) generate unique conformers, (ii) generate unique macrocycle ring conformations, (iii) identify the global energy minimum, (iv) identify conformers similar to the X-ray ligand conformation after Protein Preparation Wizard treatment (X-ray), and (v) to the X-ray ring conformation. Computational speed was also considered. In addition, conformational coverage, as defined by the number of conformations identified, was studied. In order to study the relative energies of the bioactive conformations, the energy differences between the global energy minima and the energy minimized X-ray structures and, the global energy minima and the MCMM-Exhaustive (1,000,000 search steps) generated conformers closest to the X-ray structure, were calculated and analysed. All searches were performed using relatively short run times (10,000 steps for MCMM, MTLMOD and MD/LLMOD). To assess the performance of the methods, they were compared to an exhaustive MCMM search using 1,000,000 search steps for each of the 44 macrocycles (requiring ca 200 times more CPU time). Prior to our analysis, we also investigated if the general search methods MCMM and MTLMOD could also be optimized for macrocycle conformational sampling. Taken together, our work concludes that the more general methods can be optimized for macrocycle modelling by slightly adjusting the settings around the ring closure bond. In most cases, MCMM and MTLMOD with either standard or enhanced settings performed well in comparison to the more specialized macrocycle sampling methods MD/LLMOD and PRIME-MCS. When using enhanced settings for MCMM and MTLMOD, the X-ray conformation was regenerated with the greatest accuracy. The, MD/LLMOD emerged as the most efficient method for generating the global energy minima.
大环化合物因其具有有趣的生物特性,是一类重要的具有药用相关性的小分子。因此,了解其构象偏好对于药物设计非常重要。鉴于大环化合物与蛋白质模型在药物发现中的重要性,我们设想对经典和最近的专门方法进行系统研究,将为该领域的其他从业者提供指导。在这项研究中,我们比较了通用的、成熟的构象分析方法蒙特卡罗多步极小化(MCMM)和混合扭转/低模态采样(MTLMOD)与两种更新的、专门的大环化合物采样技术:MacroModel 大环化合物基线搜索(MD/LLMOD)和 Prime 大环化合物构象采样(PRIME-MCS)的性能。我们从 44 个大环化合物与蛋白质的 X 射线晶体结构复合物中提取大环化合物,基于它们生成独特构象的能力、生成独特大环化合物环构象的能力、识别全局能量最小化的能力、在经过蛋白准备向导处理(X 射线)后识别与 X 射线配体构象相似的构象的能力,以及识别与 X 射线环构象相似的构象的能力,对每种方法进行了评估。还考虑了计算速度。此外,还研究了定义为所识别构象数量的构象覆盖率。为了研究生物活性构象的相对能量,计算并分析了全局能量最小值与能量最小化 X 射线结构之间的能量差,以及全局能量最小值与最接近 X 射线结构的 MCMM-Exhaustive(1,000,000 步搜索)生成构象之间的能量差。所有搜索都使用相对较短的运行时间(MCMM、MTLMOD 和 MD/LLMOD 为 10,000 步)进行。为了评估方法的性能,我们将其与针对 44 个大环化合物中的每个大环化合物使用 1,000,000 步搜索的 MCMM-Exhaustive 搜索进行了比较(需要大约 200 倍的 CPU 时间)。在我们的分析之前,我们还研究了通用搜索方法 MCMM 和 MTLMOD 是否也可以针对大环化合物构象采样进行优化。总的来说,我们的工作得出的结论是,通过稍微调整环闭键周围的设置,可以针对大环化合物建模对更通用的方法进行优化。在大多数情况下,使用标准或增强设置的 MCMM 和 MTLMOD 与更专门的大环化合物采样方法 MD/LLMOD 和 PRIME-MCS 相比表现良好。使用 MCMM 和 MTLMOD 的增强设置时,以最大的准确性重新生成了 X 射线构象。然后,MD/LLMOD 成为生成全局能量最小值的最有效方法。