Gubra, Hørsholm Kongevej 11B, Hørsholm, Denmark.
Department of Biochemistry and Molecular Biology, University of Southern Denmark, Odense, Denmark.
Sci Rep. 2020 Jan 24;10(1):1148. doi: 10.1038/s41598-020-58059-7.
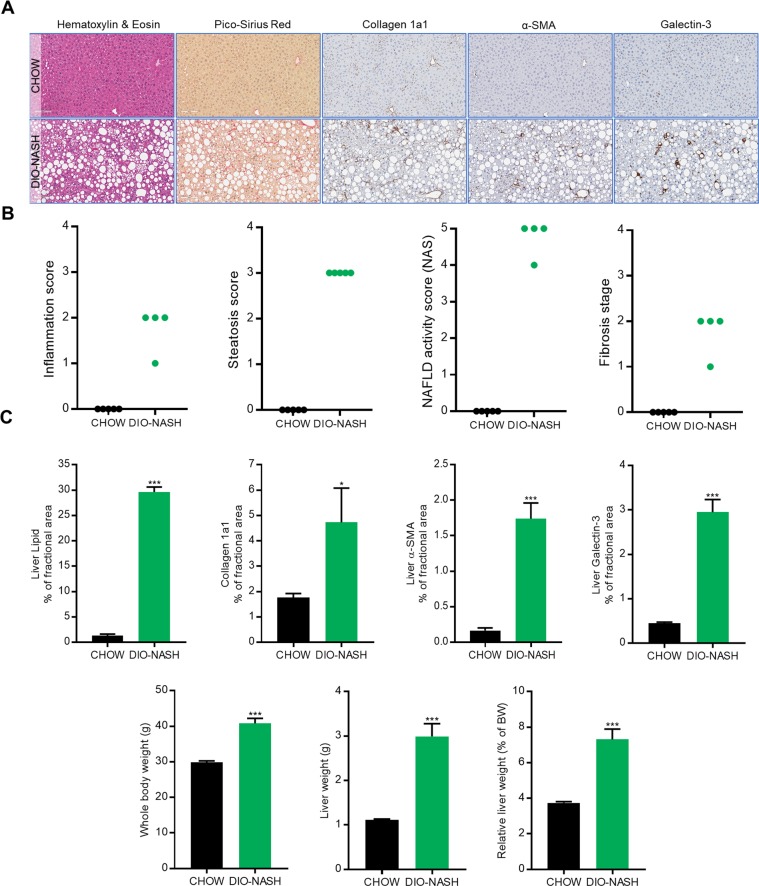
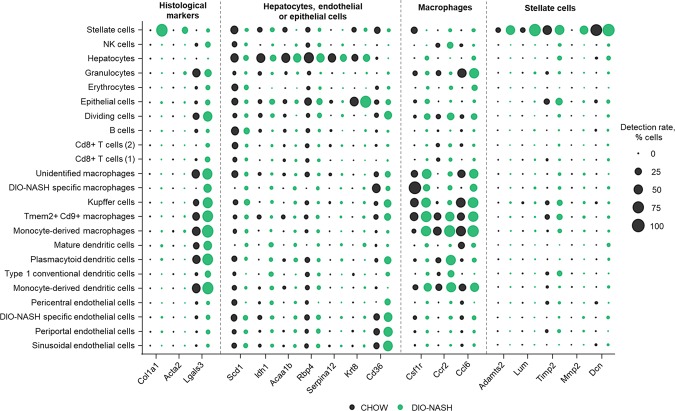
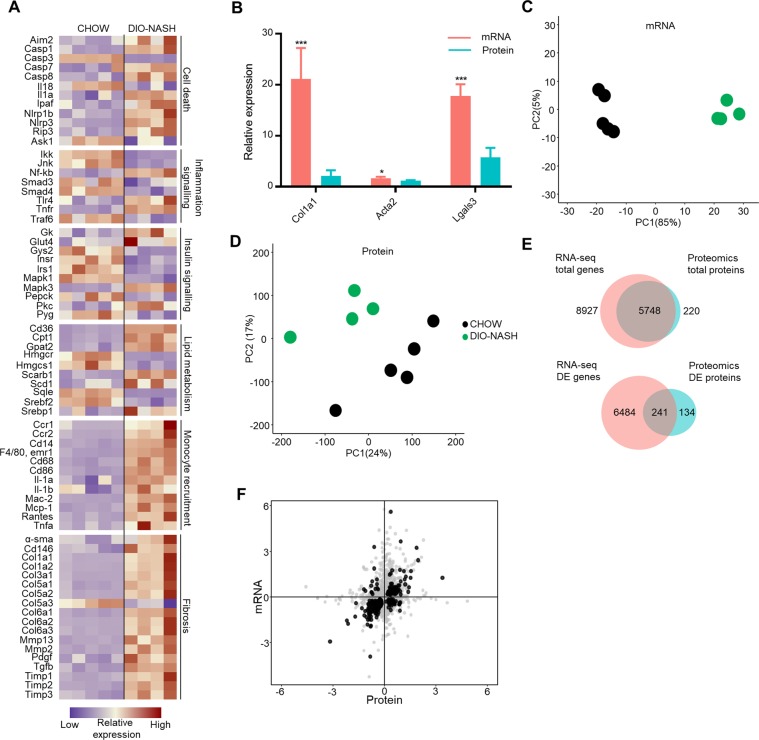
To improve the understanding of the complex biological processes underlying the development of non-alcoholic steatohepatitis (NASH), a multi-omics approach combining bulk RNA-sequencing based transcriptomics, quantitative proteomics and single-cell RNA-sequencing was used to characterize tissue biopsies from histologically validated diet-induced obese (DIO) NASH mice compared to chow-fed controls. Bulk RNA-sequencing and proteomics showed a clear distinction between phenotypes and a good correspondence between mRNA and protein level regulations, apart from specific regulatory events discovered by each technology. Transcriptomics-based gene set enrichment analysis revealed changes associated with key clinical manifestations of NASH, including impaired lipid metabolism, increased extracellular matrix formation/remodeling and pro-inflammatory responses, whereas proteomics-based gene set enrichment analysis pinpointed metabolic pathway perturbations. Integration with single-cell RNA-sequencing data identified key regulated cell types involved in development of NASH demonstrating the cellular heterogeneity and complexity of NASH pathogenesis.
为了深入了解非酒精性脂肪性肝炎(NASH)发病的复杂生物学过程,采用了一种多组学方法,结合基于批量 RNA 测序的转录组学、定量蛋白质组学和单细胞 RNA 测序,对组织活检进行了分析,这些活检来自经过组织学验证的饮食诱导肥胖(DIO)NASH 小鼠与正常饮食对照相比。批量 RNA 测序和蛋白质组学显示表型之间存在明显差异,mRNA 和蛋白质水平调控之间具有良好的一致性,除了每种技术发现的特定调控事件。基于转录组学的基因集富集分析揭示了与 NASH 的关键临床表现相关的变化,包括脂质代谢受损、细胞外基质形成/重塑和促炎反应增加,而基于蛋白质组学的基因集富集分析则指出了代谢途径的紊乱。与单细胞 RNA 测序数据的整合确定了参与 NASH 发展的关键调节细胞类型,证明了 NASH 发病机制的细胞异质性和复杂性。