Ph.D. Program in Biology, The Graduate Center, The City University of New York, New York, NY 10016, USA.
Department of Astronomy, Columbia University, New York, NY 10027, USA.
Bioinformatics. 2020 May 1;36(9):2787-2795. doi: 10.1093/bioinformatics/btaa064.
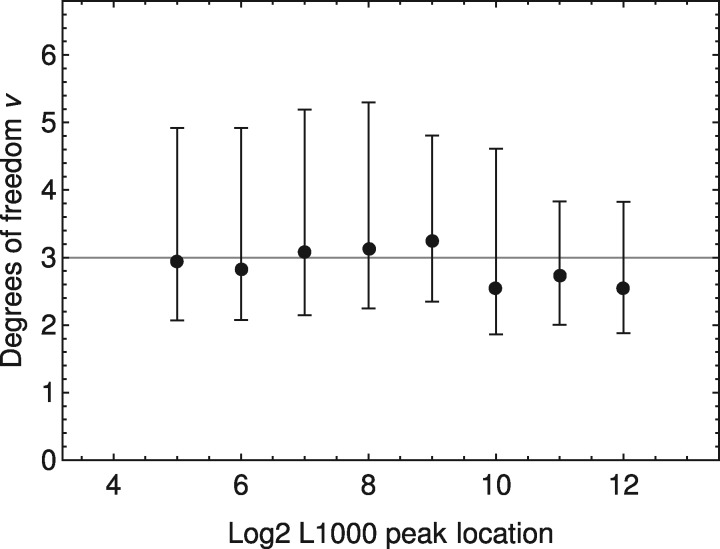
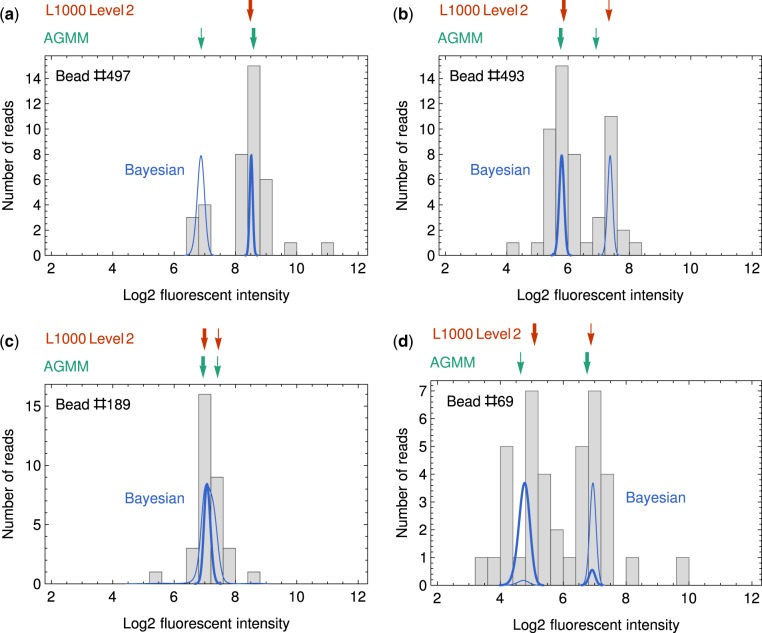
LINCS L1000 dataset contains numerous cellular expression data induced by large sets of perturbagens. Although it provides invaluable resources for drug discovery as well as understanding of disease mechanisms, the existing peak deconvolution algorithms cannot recover the accurate expression level of genes in many cases, inducing severe noise in the dataset and limiting its applications in biomedical studies.
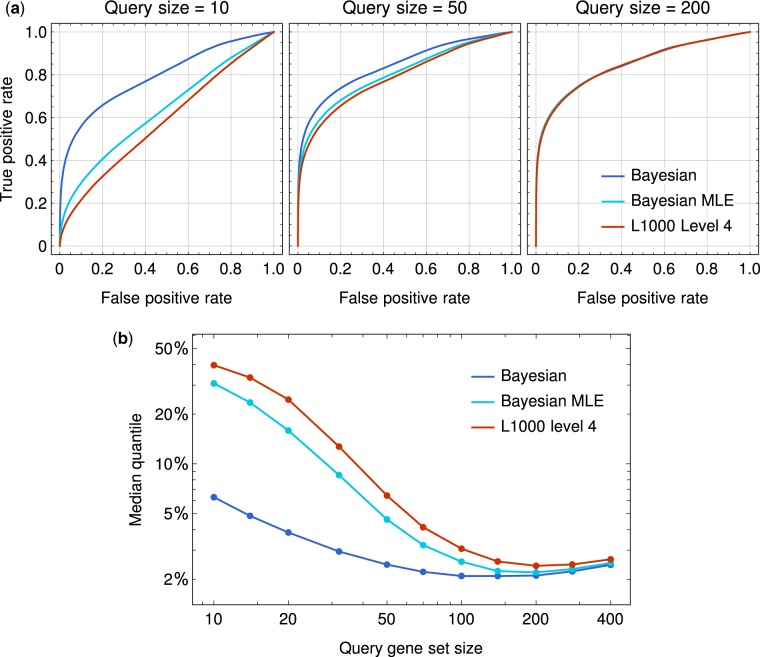
Here, we present a novel Bayesian-based peak deconvolution algorithm that gives unbiased likelihood estimations for peak locations and characterize the peaks with probability based z-scores. Based on the above algorithm, we build a pipeline to process raw data from L1000 assay into signatures that represent the features of perturbagen. The performance of the proposed pipeline is evaluated using similarity between the signatures of bio-replicates and the drugs with shared targets, and the results show that signatures derived from our pipeline gives a substantially more reliable and informative representation for perturbagens than existing methods. Thus, the new pipeline may significantly boost the performance of L1000 data in the downstream applications such as drug repurposing, disease modeling and gene function prediction.
The code and the precomputed data for LINCS L1000 Phase II (GSE 70138) are available at https://github.com/njpipeorgan/L1000-bayesian.
Supplementary data are available at Bioinformatics online.
Lincs L1000 数据集包含大量由大量扰动剂诱导的细胞表达数据。虽然它为药物发现以及了解疾病机制提供了宝贵的资源,但现有的峰分解算法在许多情况下无法恢复基因的准确表达水平,从而在数据集中引入了严重的噪声,并限制了其在生物医学研究中的应用。
在这里,我们提出了一种新的基于贝叶斯的峰分解算法,该算法可以为峰位置提供无偏的似然估计,并使用基于概率的 z 分数来描述峰的特征。基于上述算法,我们构建了一个从 L1000 分析中处理原始数据的管道,将其转化为代表扰动剂特征的特征。使用生物重复签名之间的相似性以及具有共享靶标的药物来评估所提出的管道的性能,结果表明,与现有方法相比,我们的管道从 L1000 数据中提取的特征签名提供了更可靠和信息更丰富的表示。因此,新的管道可能会显著提高 L1000 数据在下游应用(如药物重定位、疾病建模和基因功能预测)中的性能。
Lincs L1000 阶段 II(GSE70138)的代码和预先计算的数据可在 https://github.com/njpipeorgan/L1000-bayesian 上获得。
补充数据可在生物信息学在线获得。