Center for Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha, Hunan Province, 410083, People's Republic of China.
Institute for Systems Biology, Seattle, WA, 98109, USA.
BMC Genomics. 2020 Feb 6;21(1):128. doi: 10.1186/s12864-020-6541-0.
Intron retention (IR) has been traditionally overlooked as 'noise' and received negligible attention in the field of gene expression analysis. In recent years, IR has become an emerging field for interrogating transcriptomes because it has been recognized to carry out important biological functions such as gene expression regulation and it has been found to be associated with complex diseases such as cancers. However, methods for detecting IR today are limited. Thus, there is a need to develop novel methods to improve IR detection.
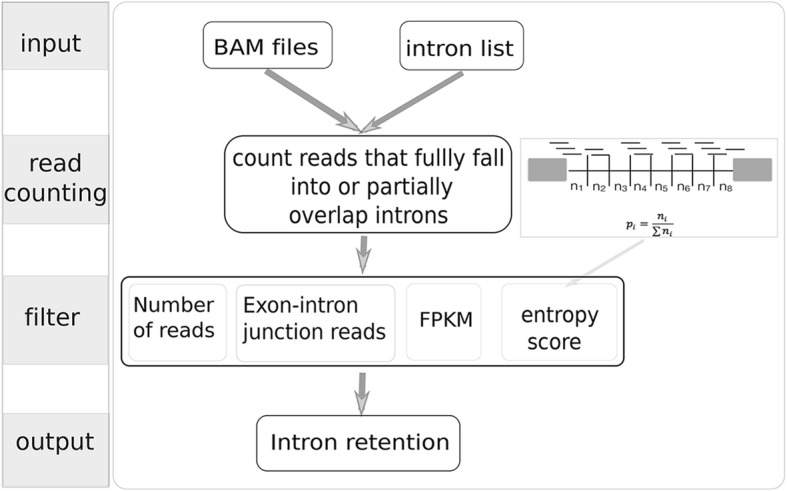
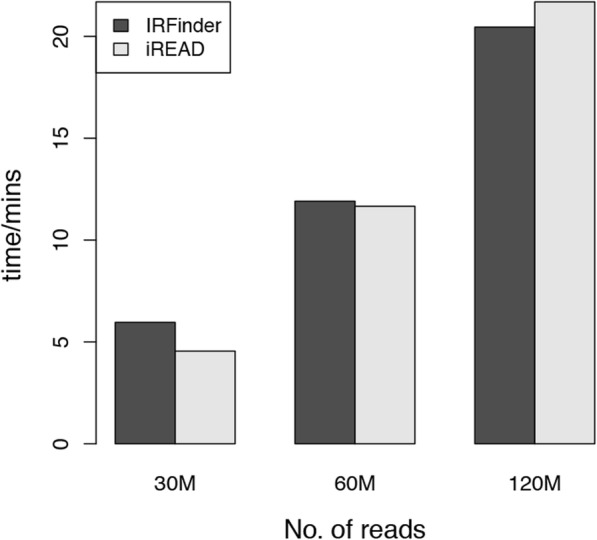
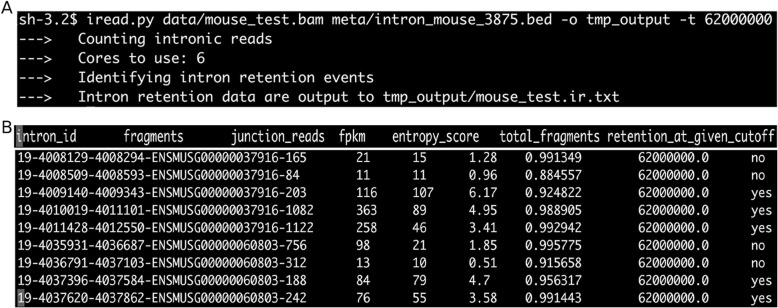
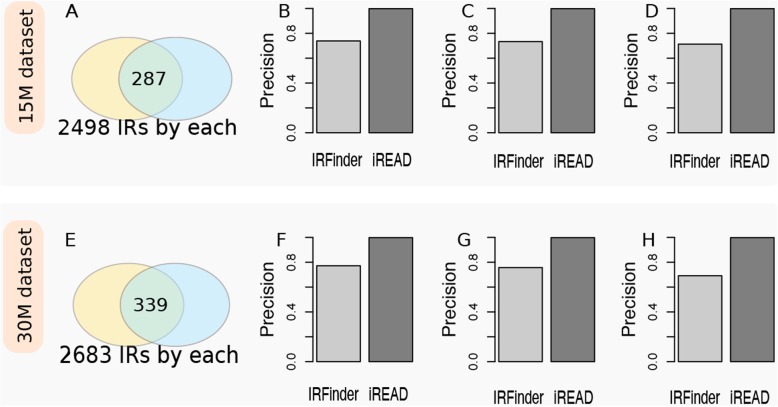
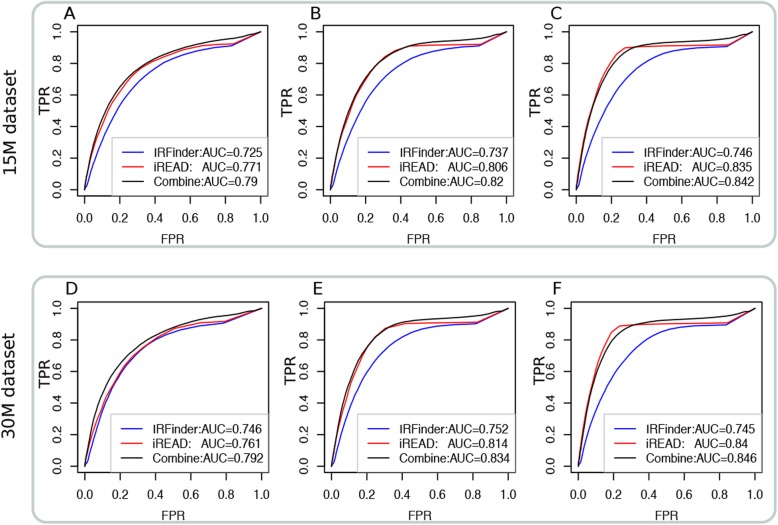
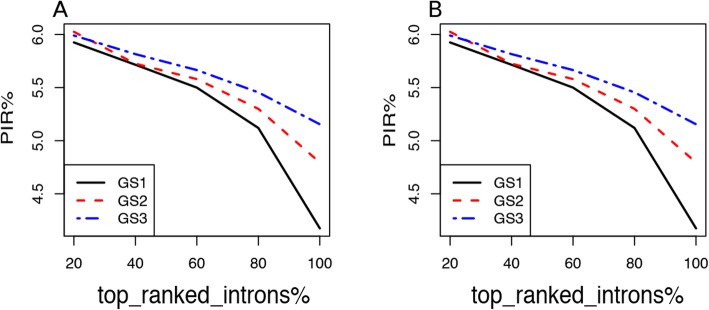
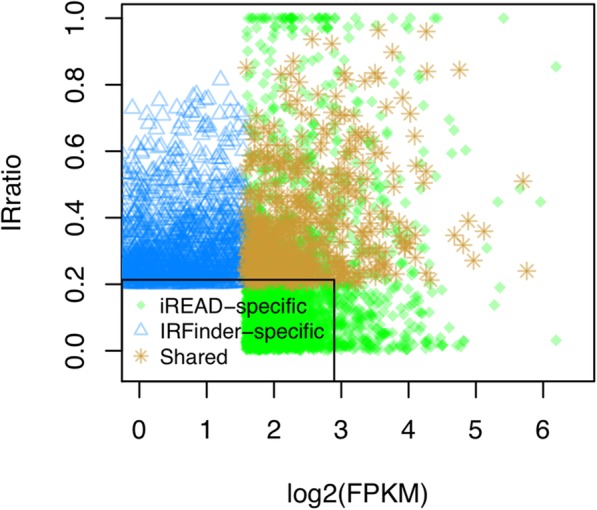
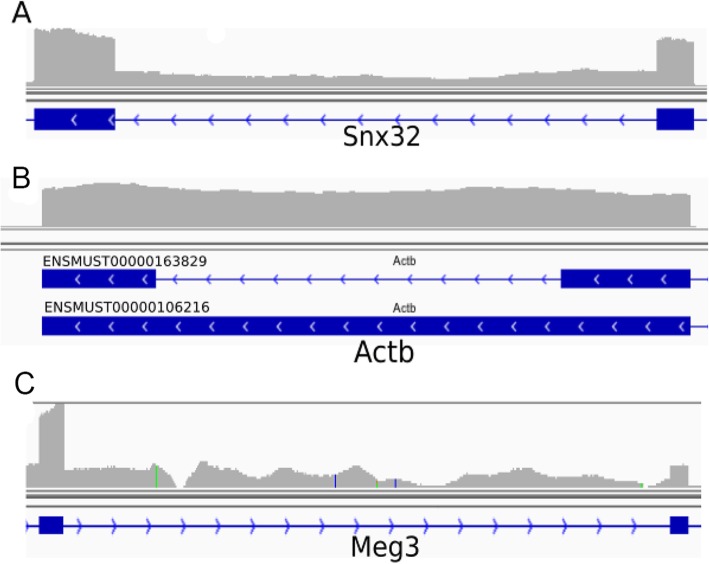
Here we present iREAD (intron REtention Analysis and Detector), a tool to detect IR events genome-wide from high-throughput RNA-seq data. The command line interface for iREAD is implemented in Python. iREAD takes as input a BAM file, representing the transcriptome, and a text file containing the intron coordinates of a genome. It then 1) counts all reads that overlap intron regions, 2) detects IR events by analyzing the features of reads such as depth and distribution patterns, and 3) outputs a list of retained introns into a tab-delimited text file. iREAD provides significant added value in detecting IR compared with output from IRFinder with a higher AUC on all datasets tested. Both methods showed low false positive rates and high false negative rates in different regimes, indicating that use together is generally beneficial. The output from iREAD can be directly used for further exploratory analysis such as differential intron expression and functional enrichment. The software is freely available at https://github.com/genemine/iread.
Being complementary to existing tools, iREAD provides a new and generic tool to interrogate poly-A enriched transcriptomic data of intron regions. Intron retention analysis provides a complementary approach for understanding transcriptome.
内含子保留(IR)传统上被视为“噪音”,在基因表达分析领域几乎没有受到关注。近年来,IR 已成为研究转录组的一个新兴领域,因为人们认识到它可以发挥重要的生物学功能,如基因表达调控,并且与癌症等复杂疾病有关。然而,目前检测 IR 的方法有限。因此,需要开发新的方法来提高 IR 检测的准确性。
我们在这里提出了 iREAD(内含子保留分析和检测),这是一种从高通量 RNA-seq 数据中全面检测 IR 事件的工具。iREAD 的命令行接口是用 Python 实现的。iREAD 以 BAM 文件(代表转录组)和包含基因组内含子坐标的文本文件作为输入。然后,它 1)计算所有与内含子区域重叠的reads;2)通过分析reads 的特征,如深度和分布模式,来检测 IR 事件;3)将保留的内含子列表输出到一个制表符分隔的文本文件中。与 IRFinder 相比,iREAD 在所有测试数据集上的 AUC 都有显著提高,在检测 IR 方面具有显著的附加价值。两种方法在不同的情况下都显示出较低的假阳性率和较高的假阴性率,这表明联合使用通常是有益的。iREAD 的输出可直接用于进一步的探索性分析,如差异内含子表达和功能富集。该软件可在 https://github.com/genemine/iread 上免费获取。
iREAD 与现有工具互补,为研究富含 poly-A 的内含子转录组数据提供了一种新的通用工具。内含子保留分析为理解转录组提供了一种补充方法。