Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, KU Leuven, 3000 Leuven, Belgium.
Spatial Epidemiology Lab (SpELL), Université Libre de Bruxelles, 1050 Brussels, Belgium.
Viruses. 2020 Feb 5;12(2):182. doi: 10.3390/v12020182.
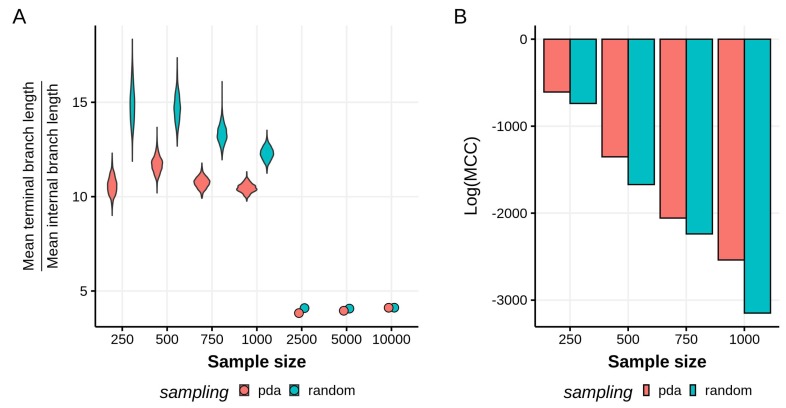
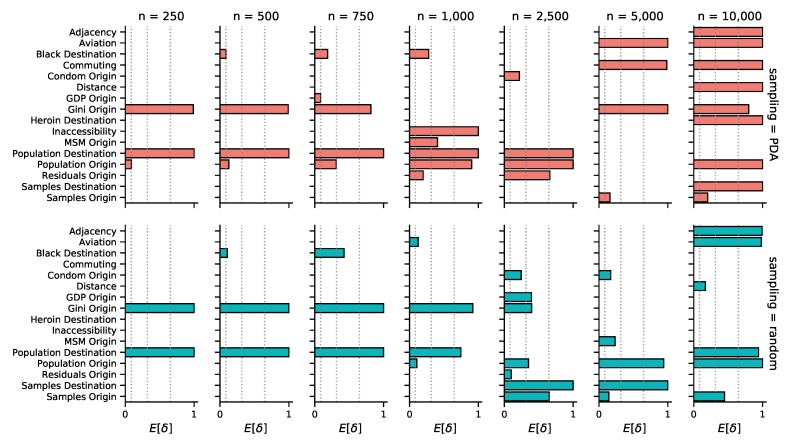
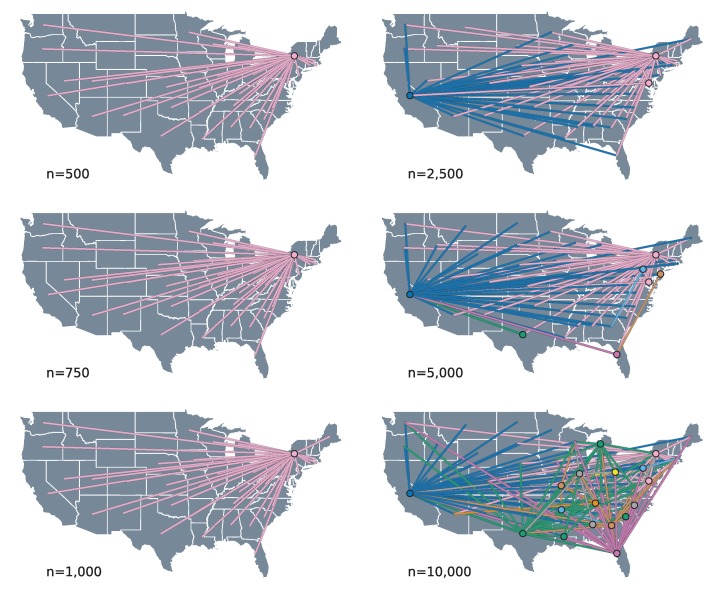
Infections with HIV-1 group M subtype B viruses account for the majority of the HIV epidemic in the Western world. Phylogeographic studies have placed the introduction of subtype B in the United States in New York around 1970, where it grew into a major source of spread. Currently, it is estimated that over one million people are living with HIV in the US and that most are infected with subtype B variants. Here, we aim to identify the drivers of HIV-1 subtype B dispersal in the United States by analyzing a collection of 23,588 ol sequences, collected for drug resistance testing from 45 states during 2004-2011. To this end, we introduce a workflow to reduce this large collection of data to more computationally-manageable sample sizes and apply the BEAST framework to test which covariates associate with the spread of HIV-1 across state borders. Our results show that we are able to consistently identify certain predictors of spread under reasonable run times across datasets of up to 10,000 sequences. However, the general lack of phylogenetic structure and the high uncertainty associated with HIV trees make it difficult to interpret the epidemiological relevance of the drivers of spread we are able to identify. While the workflow we present here could be applied to other virus datasets of a similar scale, the characteristic star-like shape of HIV-1 phylogenies poses a serious obstacle to reconstructing a detailed evolutionary and spatial history for HIV-1 subtype B in the US.
感染 HIV-1 组 M 亚型 B 病毒占西方世界艾滋病流行的大多数。系统地理学研究将 B 亚型的引入定位在美国纽约,大约在 1970 年,自此它成为了主要的传播源。目前,据估计,美国有超过 100 万人感染 HIV,其中大多数人感染的是 B 亚型变体。在这里,我们旨在通过分析 2004 年至 2011 年期间从美国 45 个州收集的用于耐药性检测的 23588 个 ol 序列,确定 HIV-1 亚型 B 在全美扩散的驱动因素。为此,我们引入了一个工作流程,以将大量数据减少到更便于计算的样本量,并应用 BEAST 框架来检验哪些协变量与 HIV-1 跨越州界的传播相关。我们的研究结果表明,我们能够在合理的运行时间内,始终如一地识别出某些传播预测因素,这些预测因素适用于最大规模为 10000 个序列的数据集。然而,由于缺乏系统发育结构以及与 HIV 树相关的高度不确定性,使得我们难以解释传播驱动因素的流行病学相关性。虽然我们在这里提出的工作流程可以应用于其他类似规模的病毒数据集,但 HIV-1 系统发育的星形特征严重阻碍了我们重建美国 HIV-1 亚型 B 的详细进化和空间历史。