Sagulenko Pavel, Puller Vadim, Neher Richard A
Max Planck Institute for Developmental Biology, Spemannstrasse 35, Tübingen 72076, Germany.
Biozentrum, University of Basel, Klingelbergstrasse 50, 4056 Basel, Switzerland.
Virus Evol. 2018 Jan 8;4(1):vex042. doi: 10.1093/ve/vex042. eCollection 2018 Jan.
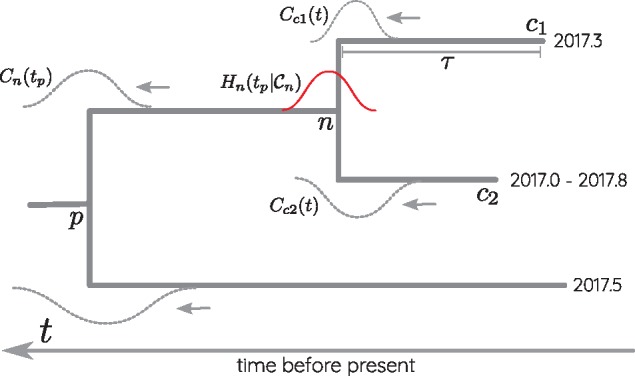
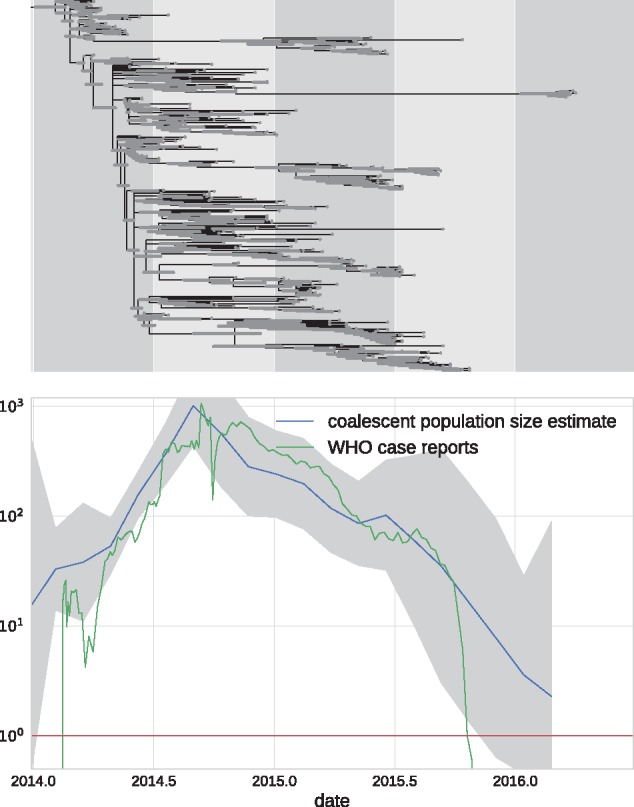
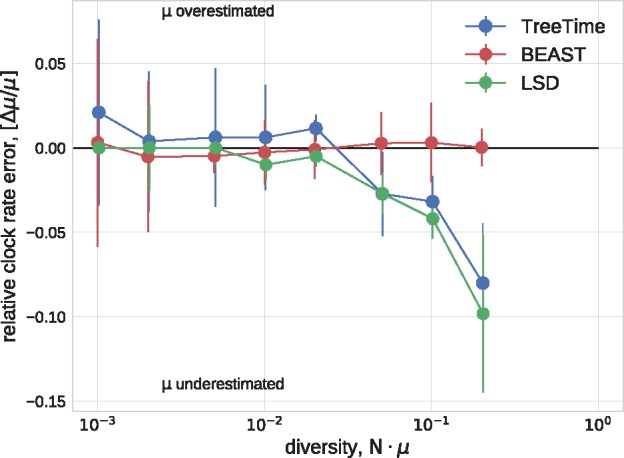
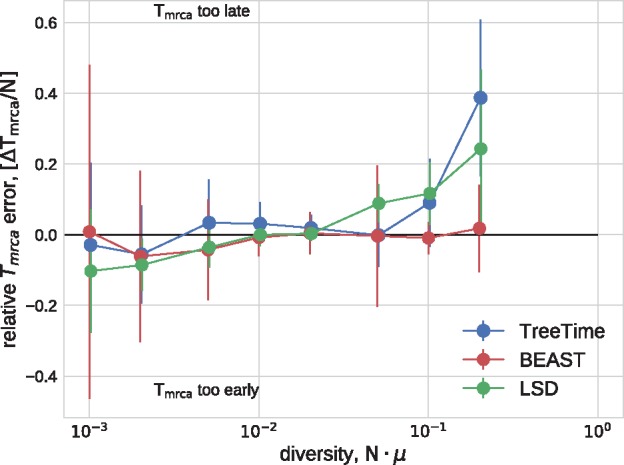
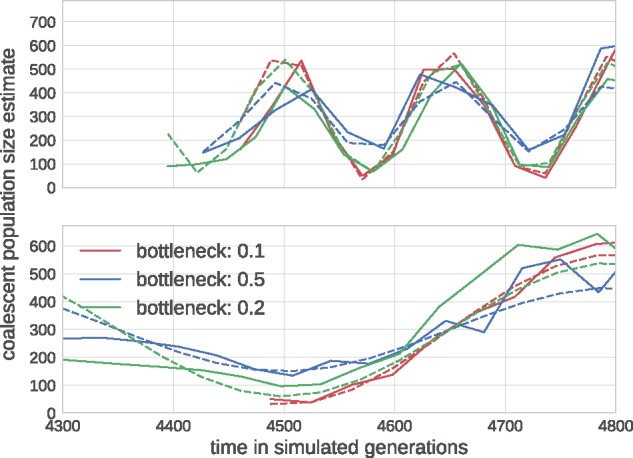
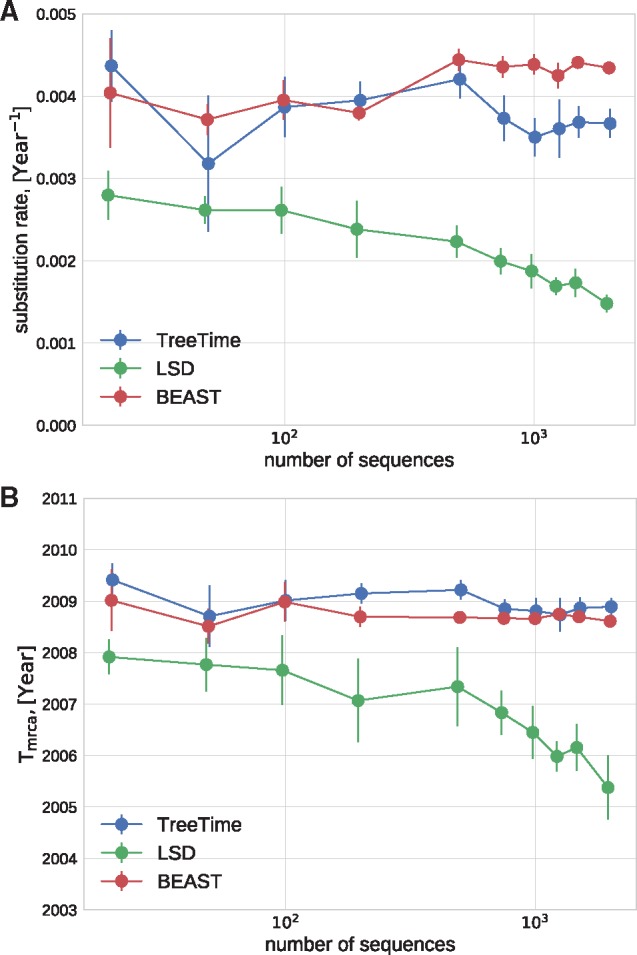
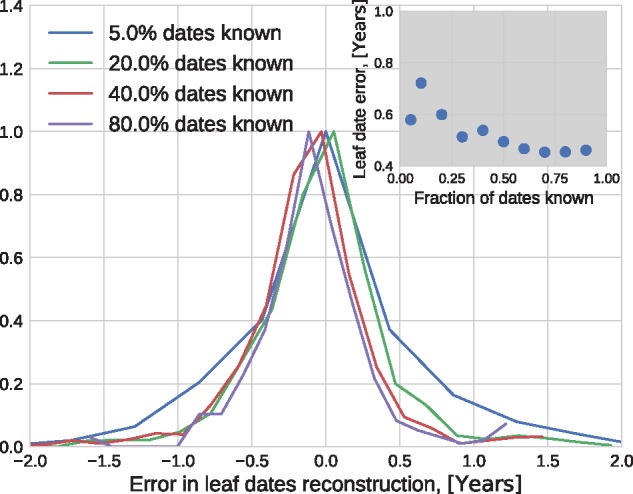
Mutations that accumulate in the genome of cells or viruses can be used to infer their evolutionary history. In the case of rapidly evolving organisms, genomes can reveal their detailed spatiotemporal spread. Such phylodynamic analyses are particularly useful to understand the epidemiology of rapidly evolving viral pathogens. As the number of genome sequences available for different pathogens has increased dramatically over the last years, phylodynamic analysis with traditional methods becomes challenging as these methods scale poorly with growing datasets. Here, we present TreeTime, a Python-based framework for phylodynamic analysis using an approximate Maximum Likelihood approach. TreeTime can estimate ancestral states, infer evolution models, reroot trees to maximize temporal signals, estimate molecular clock phylogenies and population size histories. The runtime of TreeTime scales linearly with dataset size.
细胞或病毒基因组中积累的突变可用于推断其进化历史。对于快速进化的生物体,基因组能够揭示其详细的时空传播情况。此类系统动力学分析对于理解快速进化的病毒病原体的流行病学特征尤为有用。在过去几年中,不同病原体可用的基因组序列数量急剧增加,使用传统方法进行系统动力学分析变得具有挑战性,因为这些方法在处理不断增长的数据集时扩展性较差。在此,我们展示了TreeTime,这是一个基于Python的系统动力学分析框架,使用近似最大似然法。TreeTime可以估计祖先状态、推断进化模型、重新确定树的根以最大化时间信号、估计分子钟系统发育和种群大小历史。TreeTime的运行时间与数据集大小呈线性关系。