Delft Bioinformatics Lab, University of Technology Delft, 2600GA, Delft, The Netherlands.
Bioinformatics Group, Wageningen University & Research, 6708 PB, Wageningen, The Netherlands.
Genet Sel Evol. 2020 Feb 7;52(1):4. doi: 10.1186/s12711-020-0528-9.
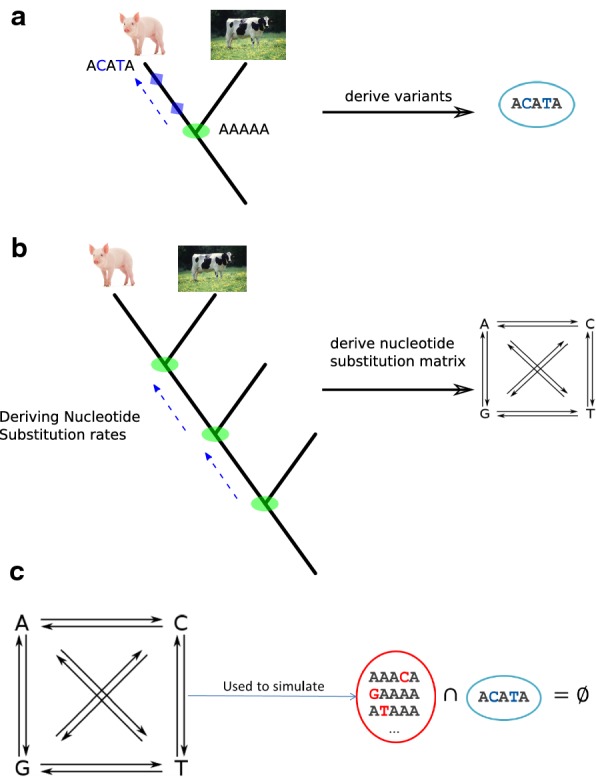
In animal breeding, identification of causative genetic variants is of major importance and high economical value. Usually, the number of candidate variants exceeds the number of variants that can be validated. One way of prioritizing probable candidates is by evaluating their potential to have a deleterious effect, e.g. by predicting their consequence. Due to experimental difficulties to evaluate variants that do not cause an amino-acid substitution, other prioritization methods are needed. For human genomes, the prediction of deleterious genomic variants has taken a step forward with the introduction of the combined annotation dependent depletion (CADD) method. In theory, this approach can be applied to any species. Here, we present pCADD (p for pig), a model to score single nucleotide variants (SNVs) in pig genomes.
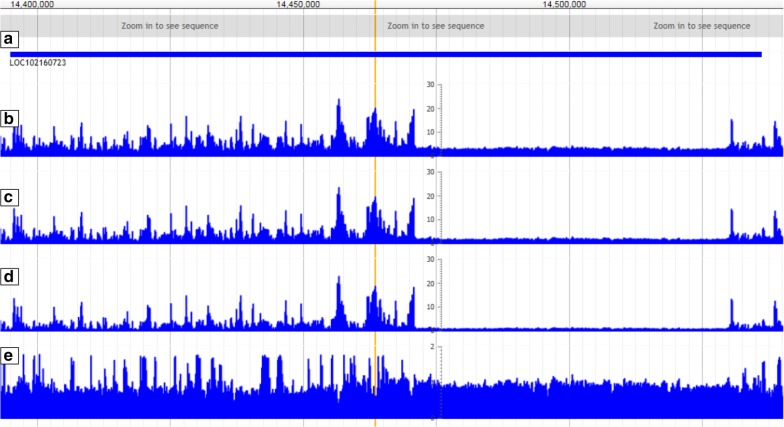
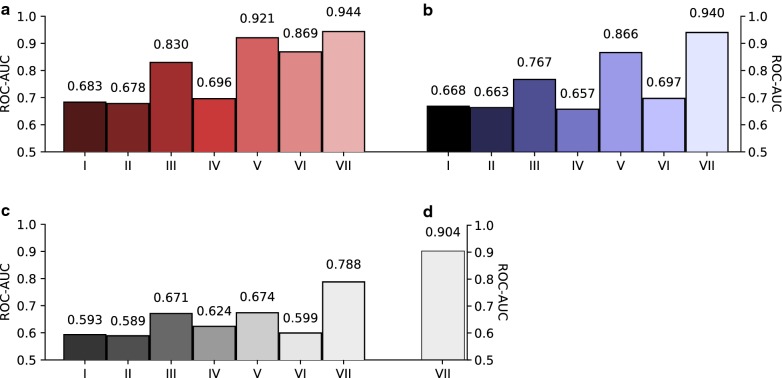
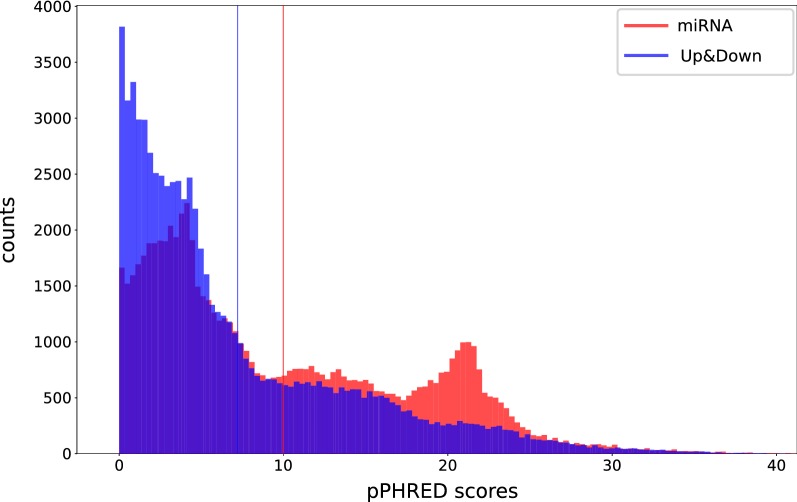
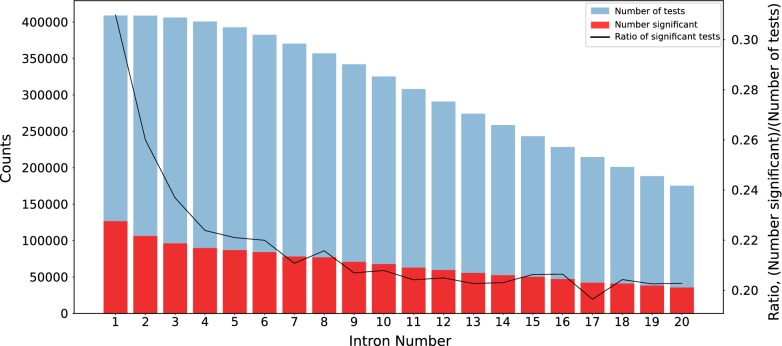
To evaluate whether pCADD captures sites with biological meaning, we used transcripts from miRNAs and introns, sequences from genes that are specific for a particular tissue, and the different sites of codons, to test how well pCADD scores differentiate between functional and non-functional elements. Furthermore, we conducted an assessment of examples of non-coding and coding SNVs, which are causal for changes in phenotypes. Our results show that pCADD scores discriminate between functional and non-functional sequences and prioritize functional SNVs, and that pCADD is able to score the different positions in a codon relative to their redundancy. Taken together, these results indicate that based on pCADD scores, regions with biological relevance can be identified and distinguished according to their rate of adaptation.
We present the ability of pCADD to prioritize SNVs in the pig genome with respect to their putative deleteriousness, in accordance to the biological significance of the region in which they are located. We created scores for all possible SNVs, coding and non-coding, for all autosomes and the X chromosome of the pig reference sequence Sscrofa11.1, proposing a toolbox to prioritize variants and evaluate sequences to highlight new sites of interest to explain biological functions that are relevant to animal breeding.
在动物育种中,鉴定致病遗传变异体具有重要的意义和巨大的经济价值。通常,候选变异体的数量超过了可验证的变异体数量。一种优先考虑可能候选者的方法是评估它们具有有害影响的潜力,例如通过预测其后果。由于评估不引起氨基酸取代的变异体的实验困难,因此需要其他优先级方法。对于人类基因组,引入综合注释依赖性耗竭(CADD)方法使有害基因组变异体的预测向前迈进了一步。从理论上讲,这种方法可以应用于任何物种。在这里,我们提出了 pCADD(p 代表猪),这是一种在猪基因组中评分单核苷酸变异(SNV)的模型。
为了评估 pCADD 是否捕获具有生物学意义的位点,我们使用了来自 miRNA 和内含子的转录本、特定于特定组织的基因的序列以及密码子的不同位点,以测试 pCADD 评分如何区分功能和非功能元件。此外,我们对导致表型变化的非编码和编码 SNV 进行了评估示例。我们的结果表明,pCADD 评分可区分功能和非功能序列,并优先考虑功能 SNV,并且 pCADD 能够相对于其冗余度评分密码子的不同位置。总之,这些结果表明,基于 pCADD 评分,可以根据其适应率识别和区分具有生物学相关性的区域。
我们展示了 pCADD 根据它们所在区域的生物学意义,对猪基因组中的 SNV 进行优先级排序的能力,以确定其潜在的有害性。我们为猪参考序列 Sscrofa11.1 的所有可能的 SNV、编码和非编码、所有常染色体和 X 染色体创建了评分,提出了一个工具包来优先考虑变体并评估序列,以突出新的感兴趣的位点,以解释与动物育种相关的生物学功能。