Department of Neurology and Neurosurgery, Montreal Neurological Institute, McGill University, 3801 University street, H3A 2B4, Montreal, Quebec, Canada.
Acta Neuropathol Commun. 2020 Feb 7;8(1):14. doi: 10.1186/s40478-020-0878-0.
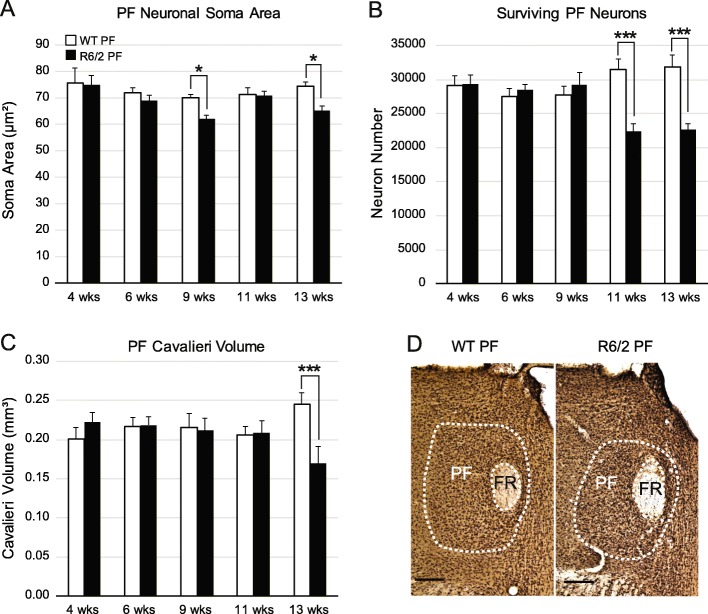
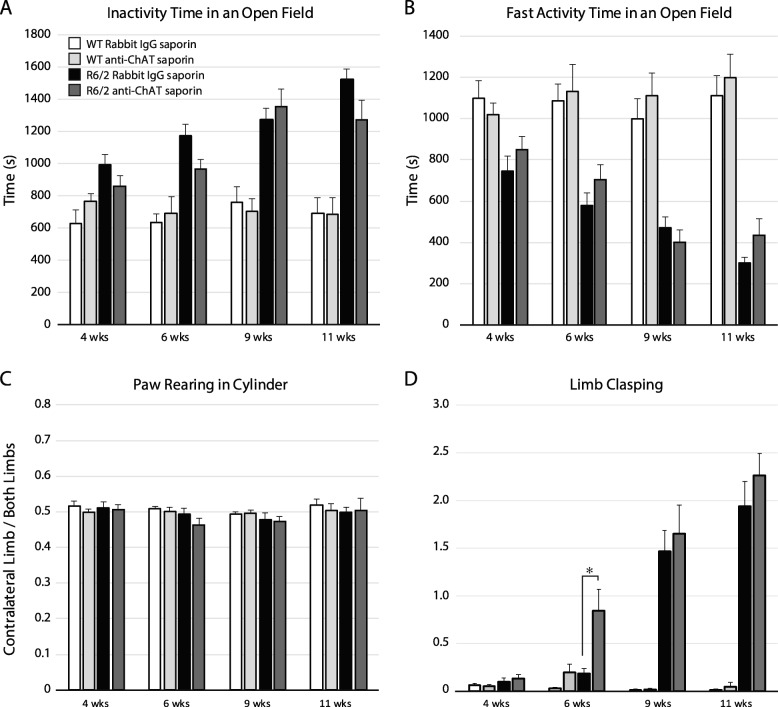
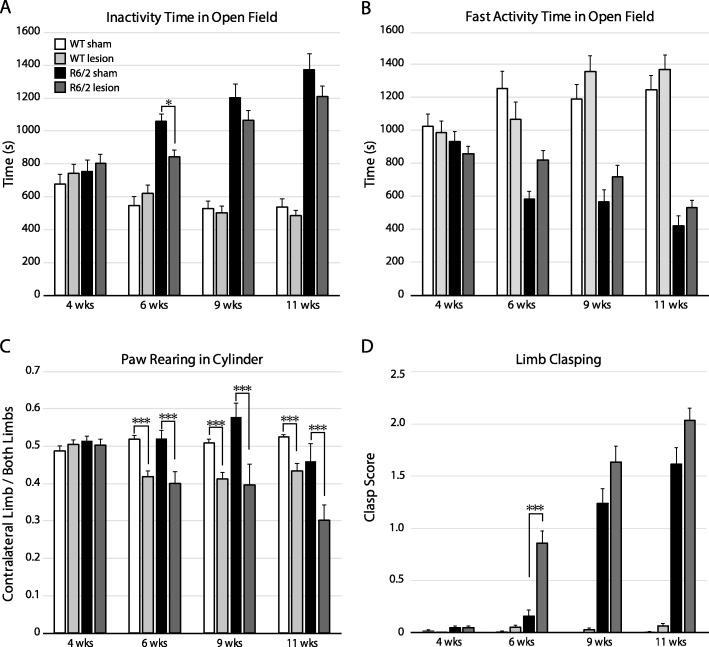
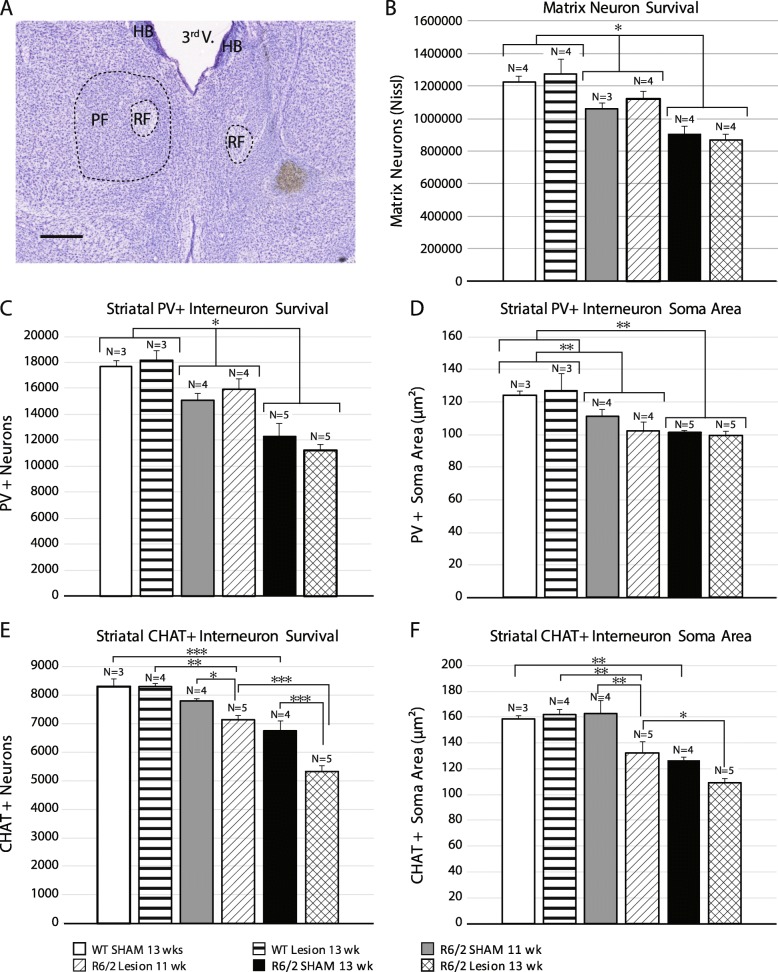
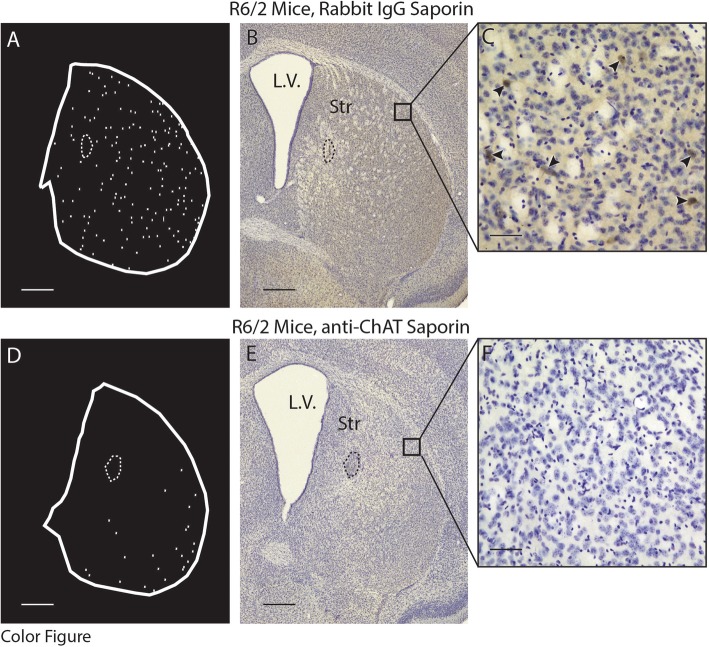
Huntington's disease (HD) is an autosomal dominant trinucleotide repeat disorder characterized by choreiform movements, dystonia and striatal neuronal loss. Amongst multiple cellular processes, abnormal neurotransmitter signalling and decreased trophic support from glutamatergic cortical afferents are major mechanisms underlying striatal degeneration. Recent work suggests that the thalamostriatal (TS) system, another major source of glutamatergic input, is abnormal in HD although its phenotypical significance is unknown. We hypothesized that TS dysfunction plays an important role in generating motor symptoms and contributes to degeneration of striatal neuronal subtypes. Our results using the R6/2 mouse model of HD indicate that neurons of the parafascicular nucleus (PF), the main source of TS afferents, degenerate at an early stage. PF lesions performed prior to motor dysfunction or striatal degeneration result in an accelerated dystonic phenotype and are associated with premature loss of cholinergic interneurons. The progressive loss of striatal medium spiny neurons and parvalbumin-positive interneurons observed in R6/2 mice is unaltered by PF lesions. Early striatal cholinergic ablation using a mitochondrial immunotoxin provides evidence for increased cholinergic vulnerability to cellular energy failure in R6/2 mice, and worsens the dystonic phenotype. The TS system therefore contributes to trophic support of striatal interneuron subtypes in the presence of neurodegenerative stress, and TS deafferentation may be a novel cell non-autonomous mechanism contributing to the pathogenesis of HD. Furthermore, behavioural experiments demonstrate that the TS system and striatal cholinergic interneurons are key motor-network structures involved in the pathogenesis of dystonia. This work suggests that treatments aimed at rescuing the TS system may preserve important elements of striatal structure and function and provide symptomatic relief in HD.
亨廷顿病(HD)是一种常染色体显性三核苷酸重复紊乱,其特征是舞蹈病、肌张力障碍和纹状体神经元丧失。在多种细胞过程中,异常神经递质信号和来自谷氨酸能皮质传入的营养支持减少是纹状体退化的主要机制。最近的工作表明,丘脑纹状体(TS)系统是另一个主要的谷氨酸能传入源,在 HD 中异常,尽管其表型意义尚不清楚。我们假设 TS 功能障碍在产生运动症状中起着重要作用,并导致纹状体神经元亚型的退化。我们使用 R6/2 小鼠 HD 模型的结果表明,丘脑束旁核(PF)神经元,TS 传入的主要来源,在早期退化。在运动功能障碍或纹状体退化之前进行 PF 损伤会导致加速的肌张力障碍表型,并与胆碱能中间神经元的过早丧失有关。在 R6/2 小鼠中观察到的纹状体中型棘突神经元和 Parvalbumin 阳性中间神经元的进行性丧失不受 PF 损伤的影响。使用线粒体免疫毒素对纹状体早期胆碱能消融提供了证据,表明 R6/2 小鼠中的胆碱能易感性增加,对细胞能量衰竭,并且使肌张力障碍表型恶化。因此,TS 系统有助于在神经退行性应激下对纹状体中间神经元亚型的营养支持,而 TS 去传入可能是导致 HD 发病机制的一种新的细胞非自主机制。此外,行为实验表明,TS 系统和纹状体胆碱能中间神经元是参与肌张力障碍发病机制的关键运动网络结构。这项工作表明,旨在挽救 TS 系统的治疗方法可能会保留纹状体结构和功能的重要元素,并为 HD 提供症状缓解。