Institute for Interdisciplinary Information Sciences, Tsinghua University, Beijing 100084, China.
The National Center for Drug Screening and the CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China.
Genomics Proteomics Bioinformatics. 2019 Oct;17(5):478-495. doi: 10.1016/j.gpb.2019.04.003. Epub 2020 Feb 6.
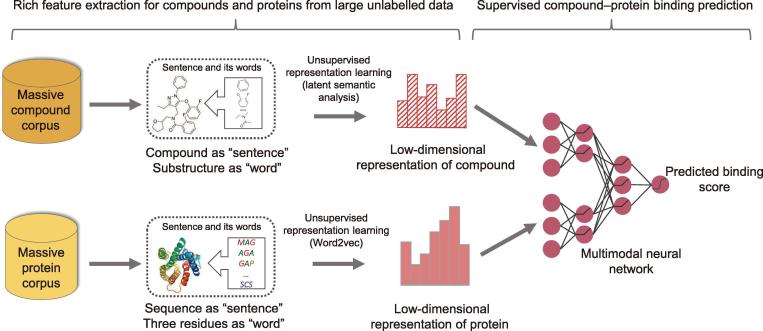
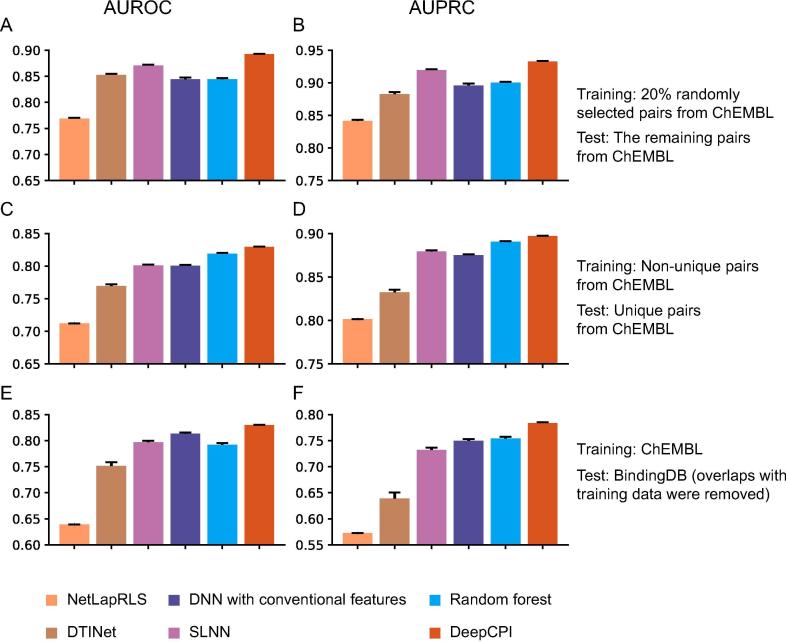
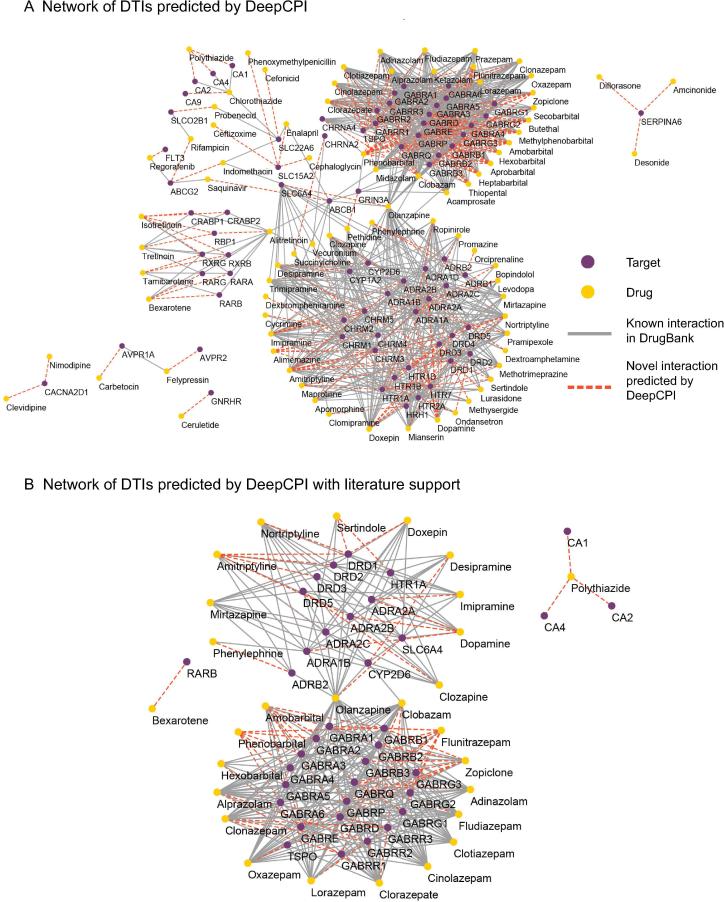
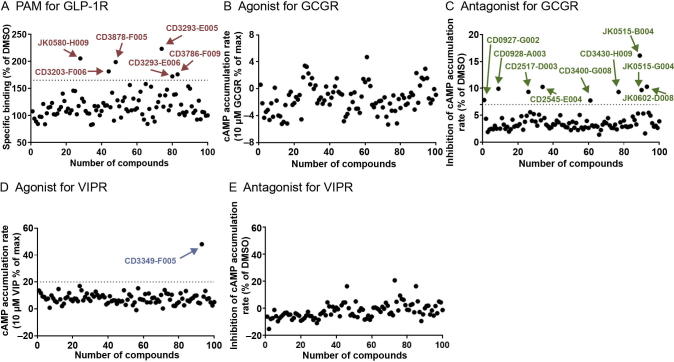
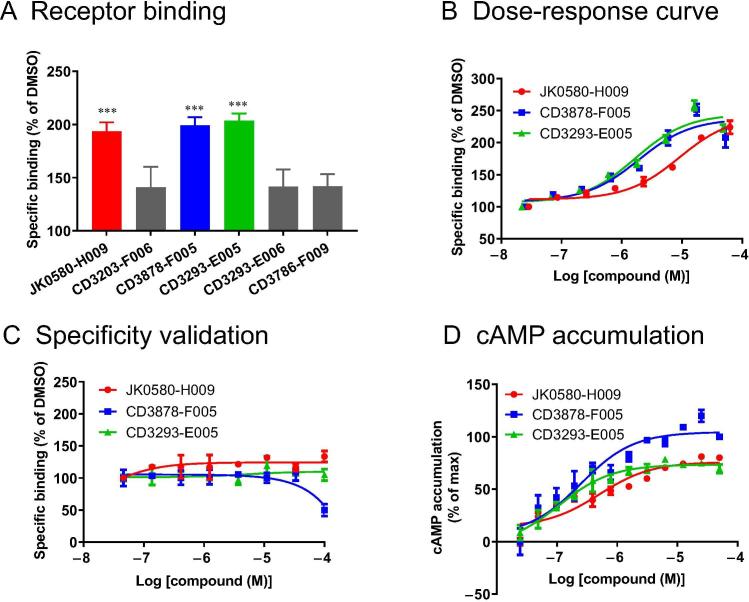
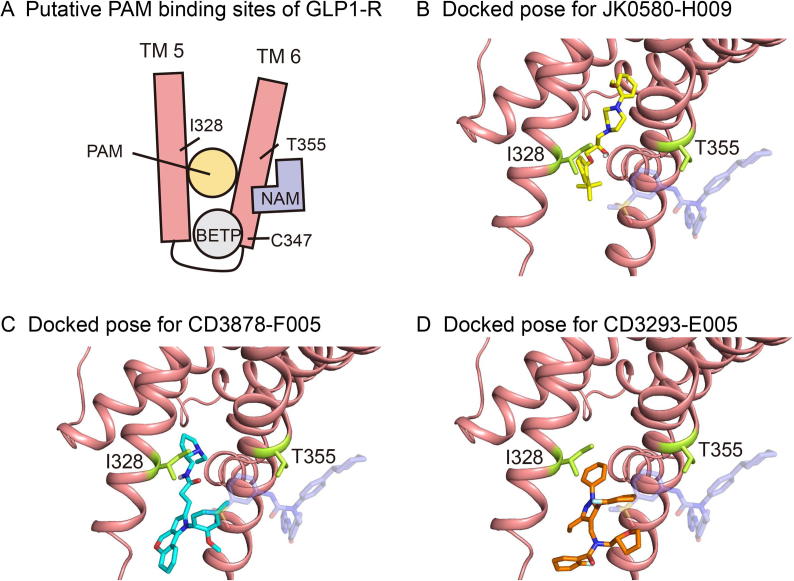
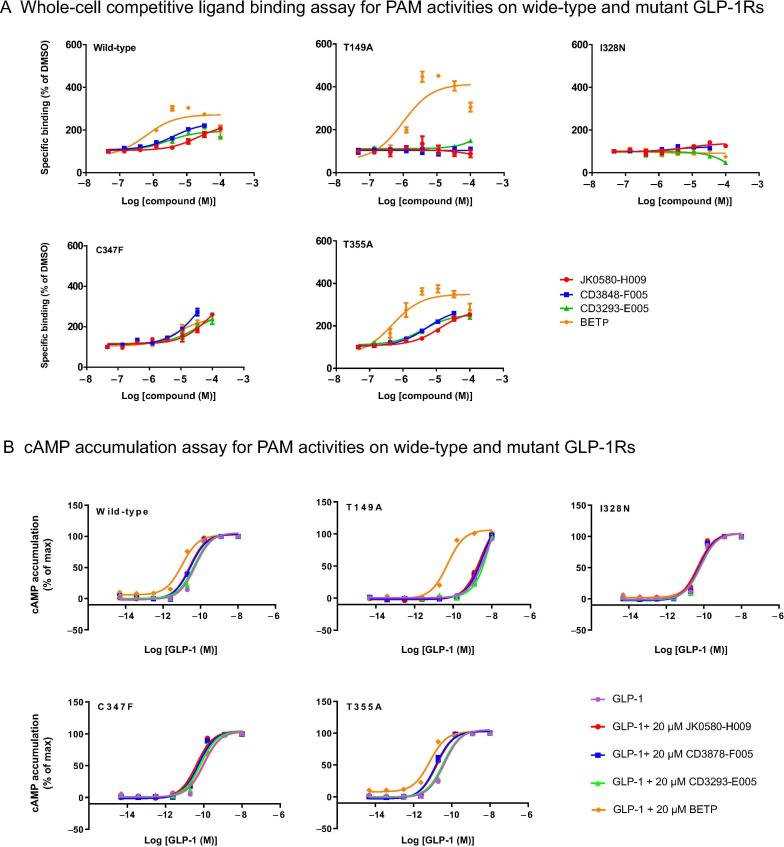
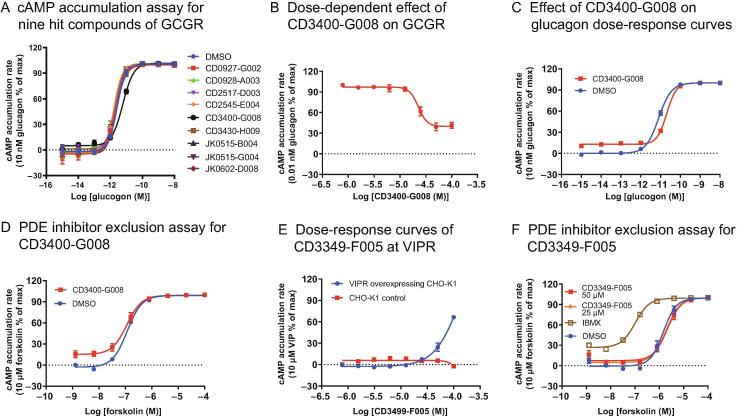
Accurate identification of compound-protein interactions (CPIs) in silico may deepen our understanding of the underlying mechanisms of drug action and thus remarkably facilitate drug discovery and development. Conventional similarity- or docking-based computational methods for predicting CPIs rarely exploit latent features from currently available large-scale unlabeled compound and protein data and often limit their usage to relatively small-scale datasets. In the present study, we propose DeepCPI, a novel general and scalable computational framework that combines effective feature embedding (a technique of representation learning) with powerful deep learning methods to accurately predict CPIs at a large scale. DeepCPI automatically learns the implicit yet expressive low-dimensional features of compounds and proteins from a massive amount of unlabeled data. Evaluations of the measured CPIs in large-scale databases, such as ChEMBL and BindingDB, as well as of the known drug-target interactions from DrugBank, demonstrated the superior predictive performance of DeepCPI. Furthermore, several interactions among small-molecule compounds and three G protein-coupled receptor targets (glucagon-like peptide-1 receptor, glucagon receptor, and vasoactive intestinal peptide receptor) predicted using DeepCPI were experimentally validated. The present study suggests that DeepCPI is a useful and powerful tool for drug discovery and repositioning. The source code of DeepCPI can be downloaded from https://github.com/FangpingWan/DeepCPI.
准确识别化合物-蛋白质相互作用(CPIs)可以深入了解药物作用的潜在机制,从而显著促进药物发现和开发。传统的基于相似性或对接的计算方法预测 CPIs 很少利用当前可用的大规模未标记化合物和蛋白质数据中的潜在特征,并且通常将其使用限制在相对较小的数据集上。在本研究中,我们提出了 DeepCPI,这是一种新颖的通用且可扩展的计算框架,它将有效的特征嵌入(一种表示学习技术)与强大的深度学习方法相结合,可大规模准确预测 CPIs。DeepCPI 可自动从大量未标记数据中学习化合物和蛋白质的隐含但有表现力的低维特征。对 ChEMBL 和 BindingDB 等大型数据库中的测量 CPIs 的评估,以及来自 DrugBank 的已知药物-靶标相互作用的评估,证明了 DeepCPI 的卓越预测性能。此外,使用 DeepCPI 预测的小分子化合物与三种 G 蛋白偶联受体靶标(胰高血糖素样肽-1 受体、胰高血糖素受体和血管活性肠肽受体)之间的几种相互作用已通过实验验证。本研究表明,DeepCPI 是一种用于药物发现和重新定位的有用且强大的工具。DeepCPI 的源代码可从 https://github.com/FangpingWan/DeepCPI 下载。