Department of Computer Engineering, Bogazici University, Istanbul, Turkey.
Department of Chemical Engineering, Bogazici University, Istanbul, Turkey.
Bioinformatics. 2018 Sep 1;34(17):i821-i829. doi: 10.1093/bioinformatics/bty593.
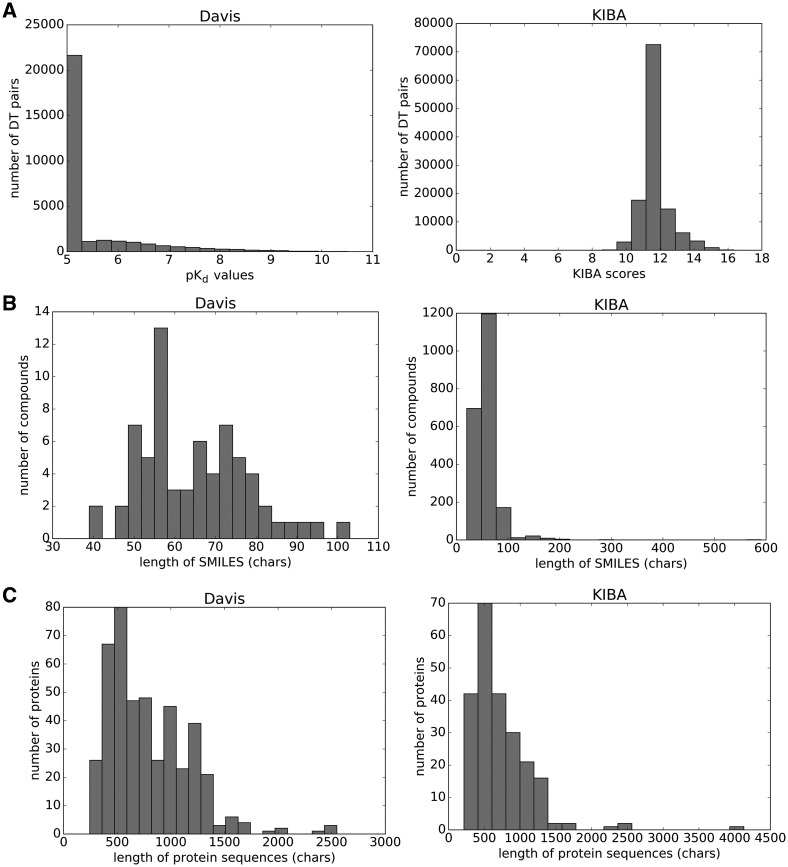
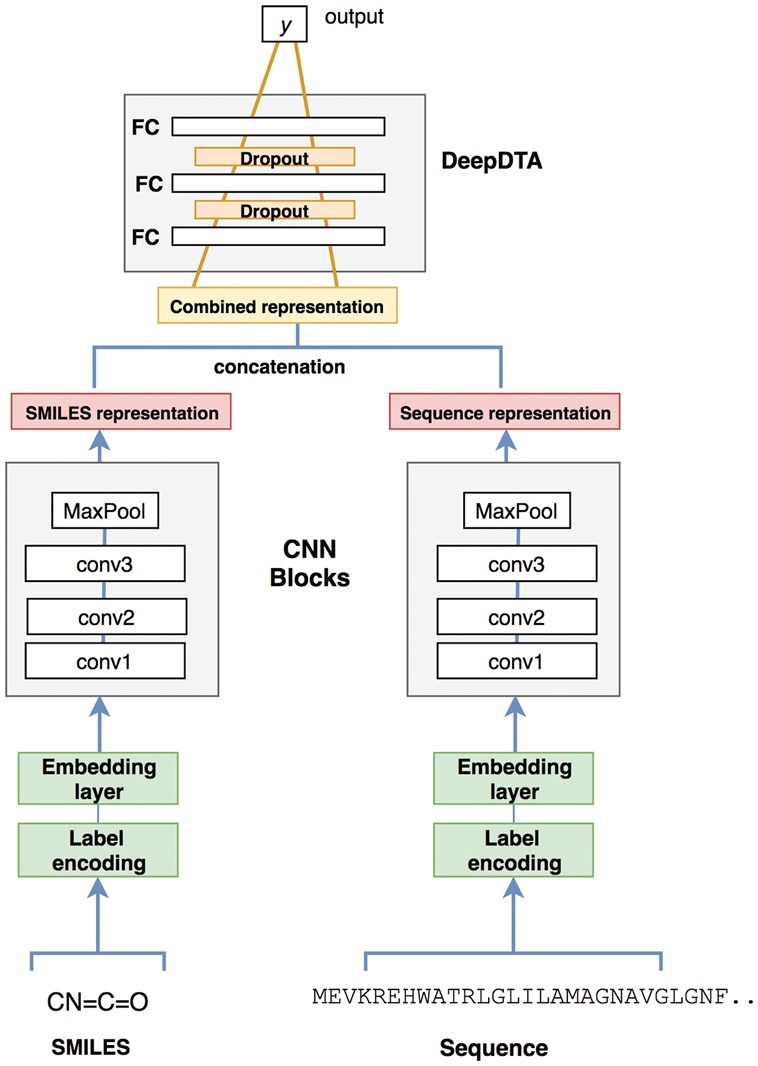
The identification of novel drug-target (DT) interactions is a substantial part of the drug discovery process. Most of the computational methods that have been proposed to predict DT interactions have focused on binary classification, where the goal is to determine whether a DT pair interacts or not. However, protein-ligand interactions assume a continuum of binding strength values, also called binding affinity and predicting this value still remains a challenge. The increase in the affinity data available in DT knowledge-bases allows the use of advanced learning techniques such as deep learning architectures in the prediction of binding affinities. In this study, we propose a deep-learning based model that uses only sequence information of both targets and drugs to predict DT interaction binding affinities. The few studies that focus on DT binding affinity prediction use either 3D structures of protein-ligand complexes or 2D features of compounds. One novel approach used in this work is the modeling of protein sequences and compound 1D representations with convolutional neural networks (CNNs).
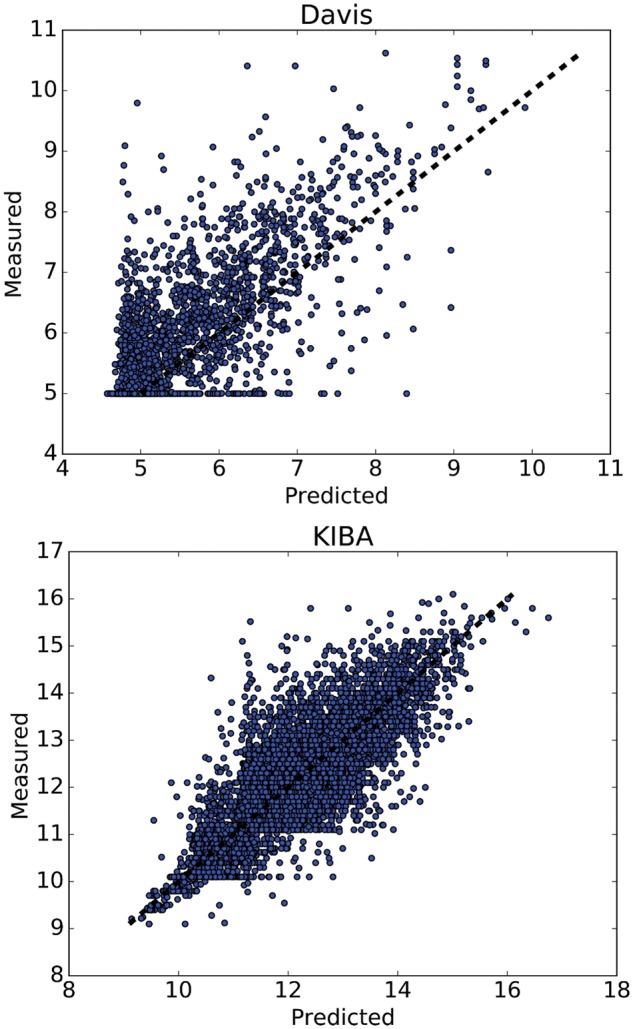
The results show that the proposed deep learning based model that uses the 1D representations of targets and drugs is an effective approach for drug target binding affinity prediction. The model in which high-level representations of a drug and a target are constructed via CNNs achieved the best Concordance Index (CI) performance in one of our larger benchmark datasets, outperforming the KronRLS algorithm and SimBoost, a state-of-the-art method for DT binding affinity prediction.
https://github.com/hkmztrk/DeepDTA.
Supplementary data are available at Bioinformatics online.
新的药物靶点 (DT) 相互作用的识别是药物发现过程的重要组成部分。大多数被提出的用于预测 DT 相互作用的计算方法都集中在二分类上,其目标是确定 DT 对是否相互作用。然而,蛋白质-配体相互作用假定为连续的结合强度值,也称为结合亲和力,预测这个值仍然是一个挑战。DT 知识库中可用的亲和力数据的增加允许在预测结合亲和力方面使用先进的学习技术,如深度学习架构。在这项研究中,我们提出了一种基于深度学习的模型,该模型仅使用目标和药物的序列信息来预测 DT 相互作用的结合亲和力。少数专注于 DT 结合亲和力预测的研究要么使用蛋白质-配体复合物的 3D 结构,要么使用化合物的 2D 特征。这项工作中使用的一种新颖方法是使用卷积神经网络 (CNN) 对蛋白质序列和化合物 1D 表示进行建模。
结果表明,使用目标和药物的 1D 表示的基于深度学习的模型是一种有效的药物靶标结合亲和力预测方法。通过 CNN 构建药物和靶标高级表示的模型在我们的一个较大的基准数据集之一中实现了最佳的一致性指数 (CI) 性能,优于 KronRLS 算法和 SimBoost,这是一种用于 DT 结合亲和力预测的最新方法。
https://github.com/hkmztrk/DeepDTA。
补充数据可在 Bioinformatics 在线获得。