Department of Pathology, LSU Health Sciences Center, Shreveport, LA, USA.
Department of Physiology and Pharmacology, West Virginia University, United States.
Redox Biol. 2020 Jul;34:101447. doi: 10.1016/j.redox.2020.101447. Epub 2020 Jan 30.
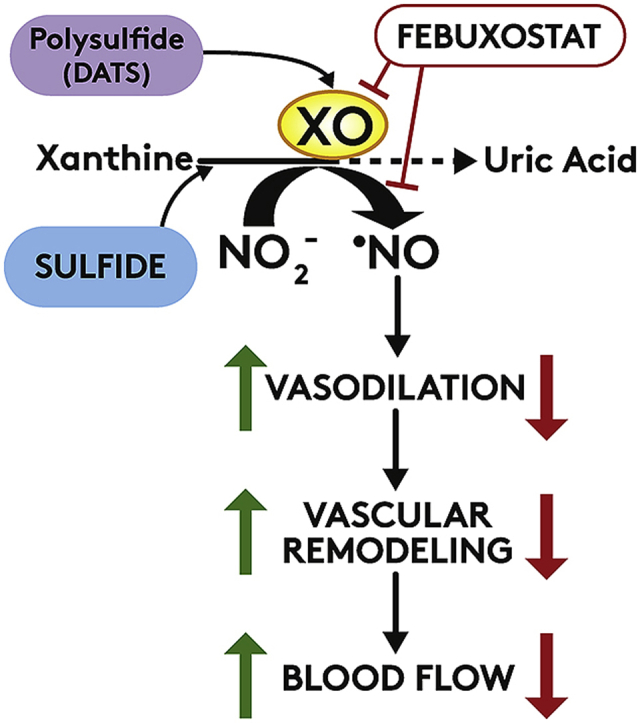
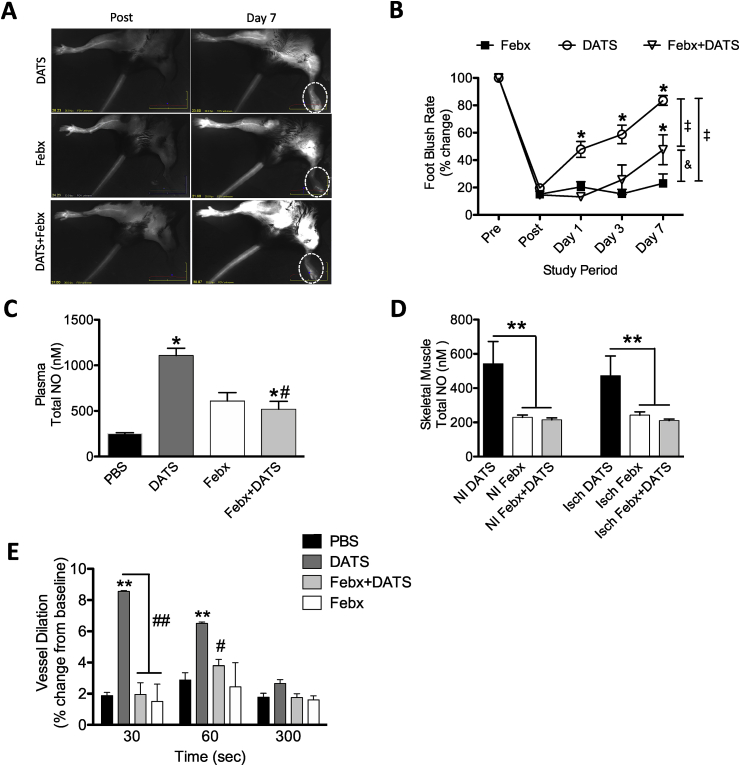
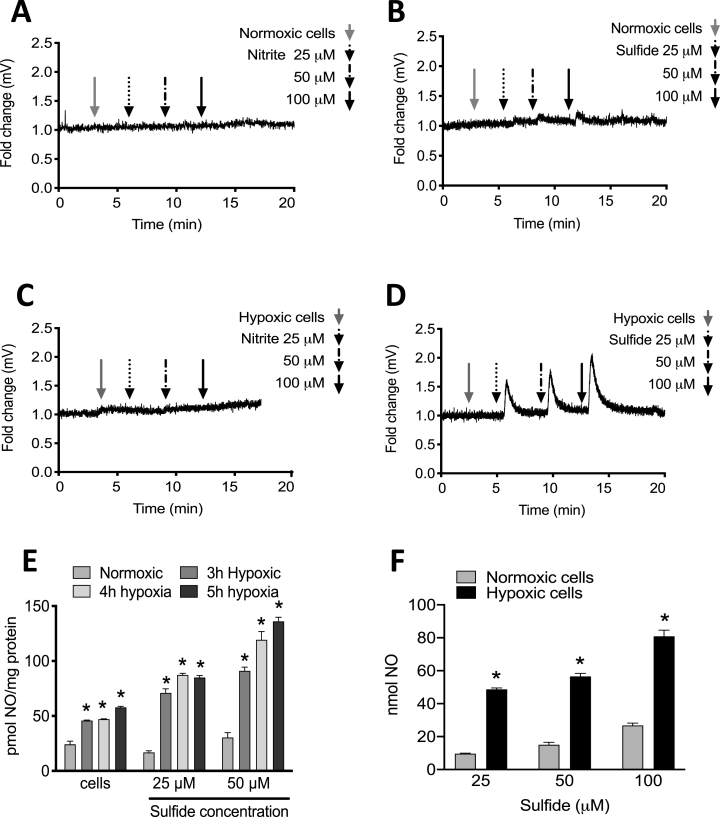
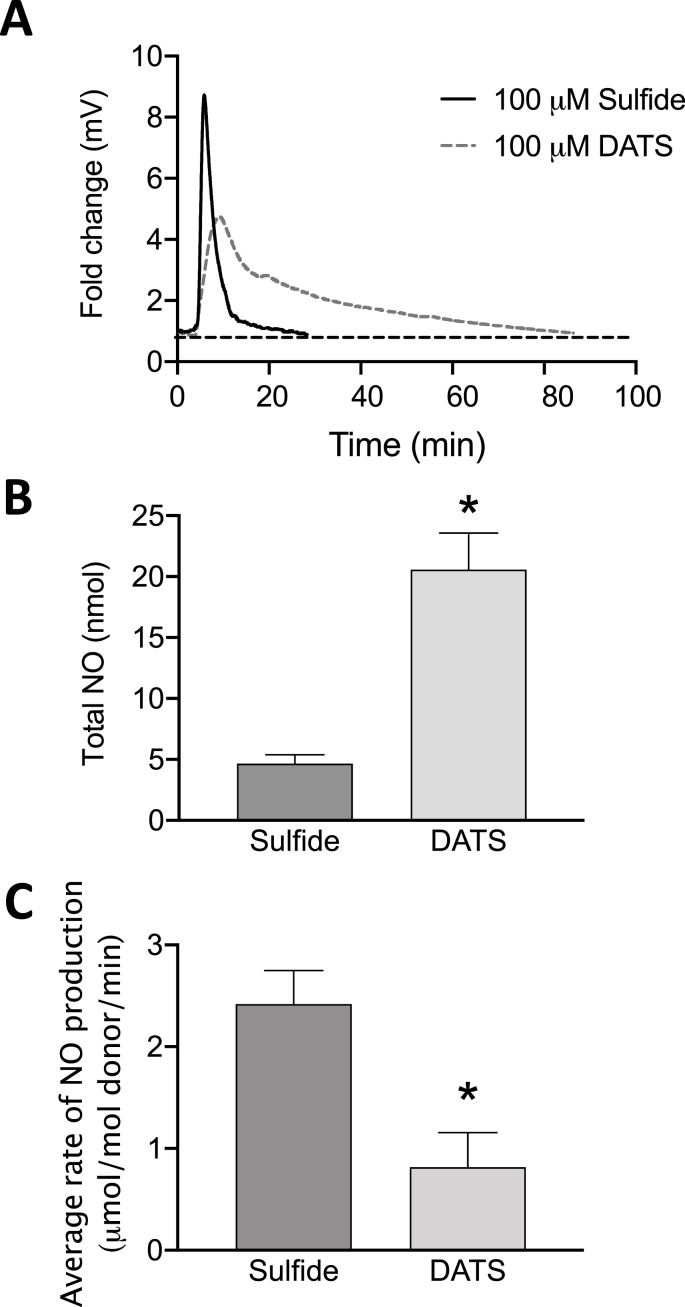
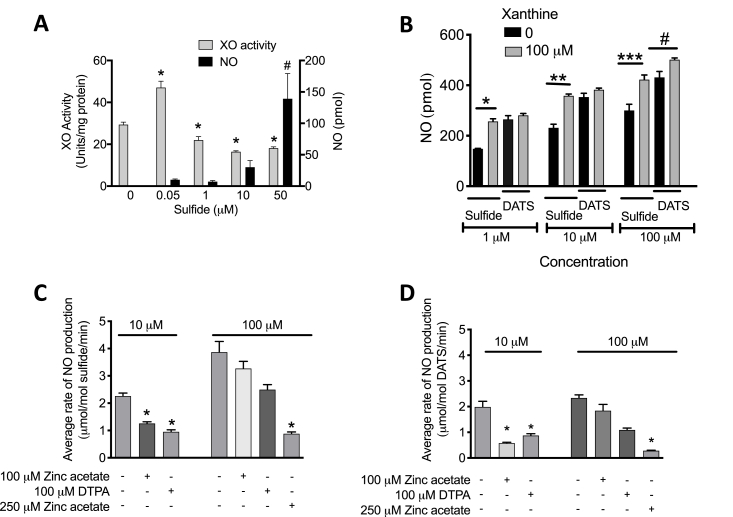
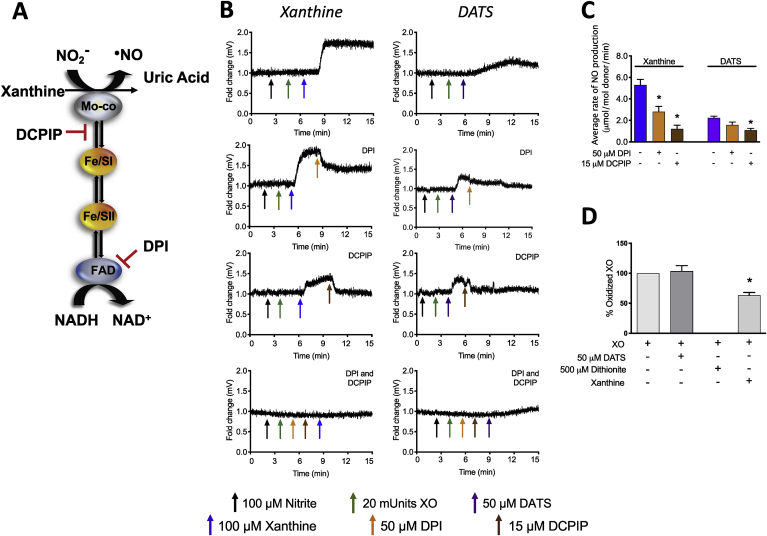
Cardiovascular disease is the leading cause of death and disability worldwide with increased oxidative stress and reduced NO bioavailability serving as key risk factors. For decades, elevation in protein abundance and enzymatic activity of xanthine oxidoreductase (XOR) under hypoxic/inflammatory conditions has been associated with organ damage and vascular dysfunction. Recent reports have challenged this dogma by identifying a beneficial function for XOR, under similar hypoxic/acidic conditions, whereby XOR catalyzes the reduction of nitrite (NO2) to nitric oxide (NO) through poorly defined mechanisms. We previously reported that hydrogen sulfide (HS/sulfide) confers significant vascular benefit under these same conditions via NO2 mediated mechanisms independent of nitric oxide synthase (NOS). Here we report for the first time the convergence of HS, XOR, and nitrite to form a concerted triad for NO generation. Specifically, hypoxic endothelial cells show a dose-dependent, sulfide and polysulfide (diallyl trisulfide (DATS)-induced, NOS-independent NO reduction to NO that is dependent upon the enzymatic activity of XOR. Interestingly, nitrite reduction to NO was found to be slower and more sustained with DATS compared to HS. Capacity for sulfide/polysulfide to produce an XOR-dependent impact on NO generation translates to salutary actions in vivo as DATS administration in cystathionine-γ-lyase (CSE) knockout mice significantly improved hindlimb ischemia blood flow post ligation, while the XOR-specific inhibitor, febuxostat (Febx), abrogated this benefit. Moreover, flow-mediated vasodilation (FMD) in CSE knockout mice following administration of DATS resulted in greater than 4-fold enhancement in femoral artery dilation while co-treatment with Febx completely completely abrogated this effect. Together, these results identify XOR as a focal point of convergence between sulfide- and nitrite-mediated signaling, as well as affirm the critical need to reexamine current dogma regarding inhibition of XOR in the context of vascular dysfunction.
心血管疾病是全球范围内导致死亡和残疾的主要原因,氧化应激增加和一氧化氮生物利用度降低是主要的风险因素。几十年来,缺氧/炎症条件下黄嘌呤氧化还原酶(XOR)的蛋白丰度和酶活性升高与器官损伤和血管功能障碍有关。最近的报道挑战了这一观点,认为在类似的缺氧/酸性条件下,XOR 具有有益的功能,通过尚未明确的机制,XOR 可以催化亚硝酸盐(NO2)转化为一氧化氮(NO)。我们之前报道过,在相同的条件下,硫化氢(HS/硫氢化物)通过 NO2 介导的机制独立于一氧化氮合酶(NOS)发挥重要的血管保护作用。在这里,我们首次报道了 HS、XOR 和亚硝酸盐在缺氧内皮细胞中形成协同三组分以产生一氧化氮。具体来说,缺氧内皮细胞显示出剂量依赖性、硫氢化物和多硫化物(二烯丙基三硫(DATS)诱导的、NOS 非依赖性的 NO 还原为 NO,这依赖于 XOR 的酶活性。有趣的是,与 HS 相比,亚硝酸盐还原为 NO 的速度较慢且更持久。硫氢化物/多硫化物产生对一氧化氮生成的 XOR 依赖性影响的能力转化为体内的有益作用,因为 DATS 在胱硫醚-γ-裂解酶(CSE)敲除小鼠中的给药显著改善了结扎后的后肢缺血血流,而 XOR 特异性抑制剂别嘌呤醇(Febx)则消除了这种益处。此外,DATS 给药后 CSE 敲除小鼠的血流介导的血管扩张(FMD)导致股动脉扩张增加了 4 倍以上,而与 Febx 共同治疗则完全消除了这种作用。总之,这些结果表明 XOR 是硫氢化物和亚硝酸盐介导的信号转导的交汇点,并证实了在血管功能障碍的背景下重新审视 XOR 抑制的当前观点的必要性。