Department of Microbial Sciences, University of Surrey, Guildford, United Kingdom.
Front Immunol. 2020 Jan 21;10:3121. doi: 10.3389/fimmu.2019.03121. eCollection 2019.
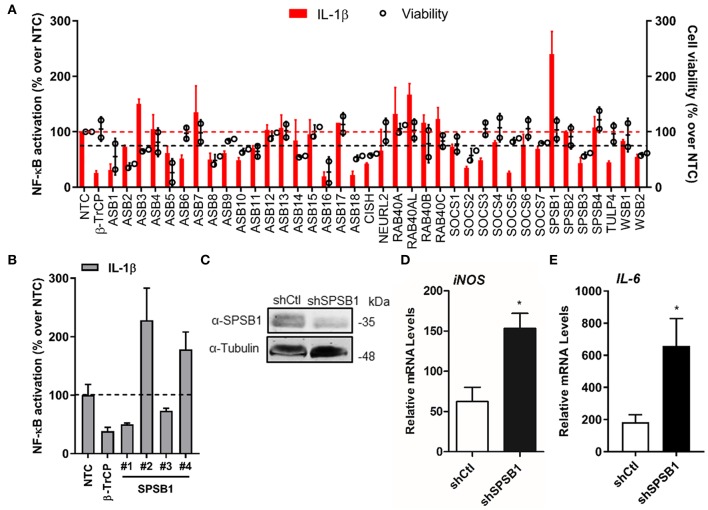
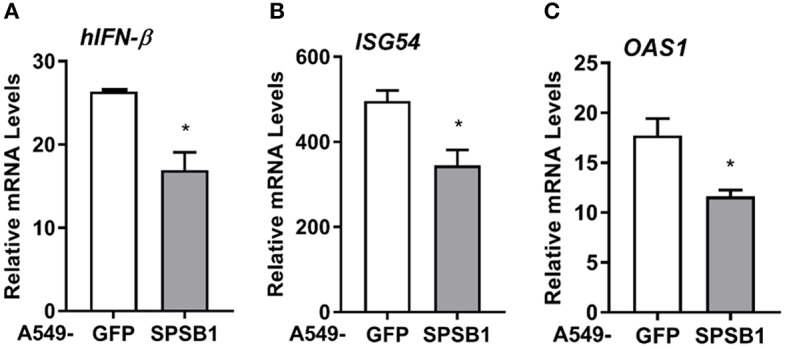
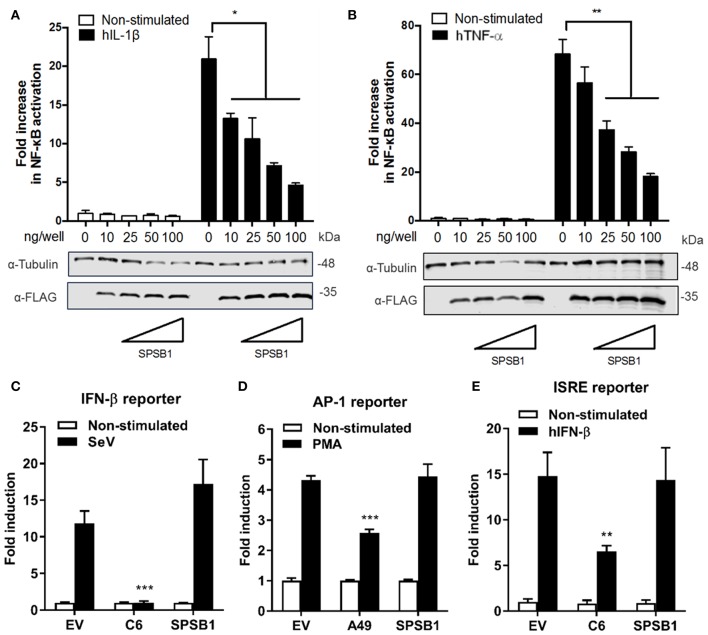
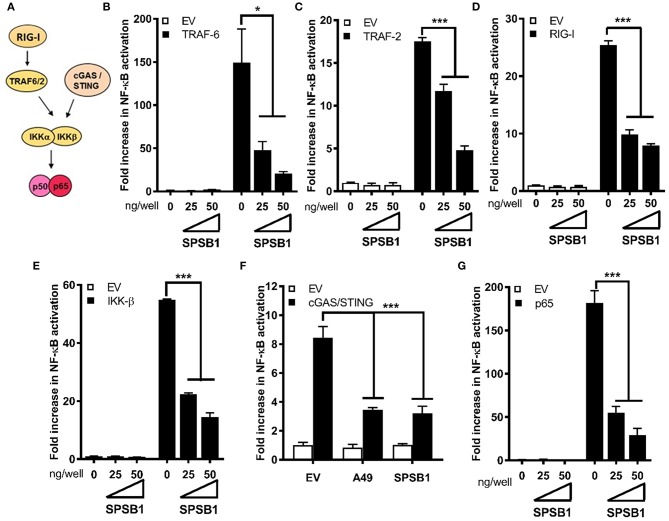
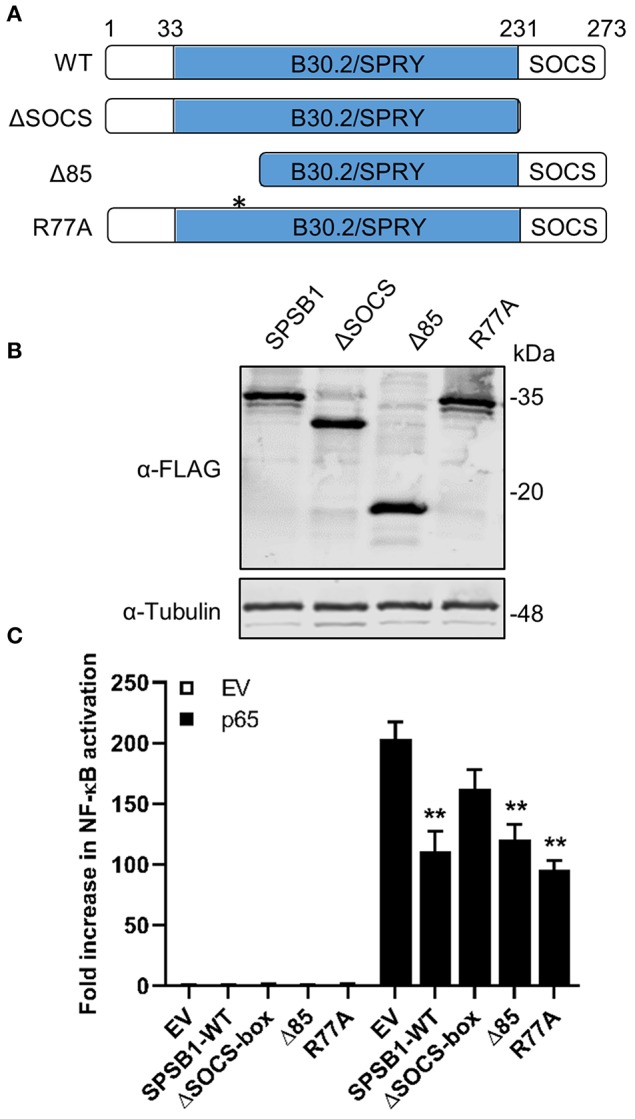
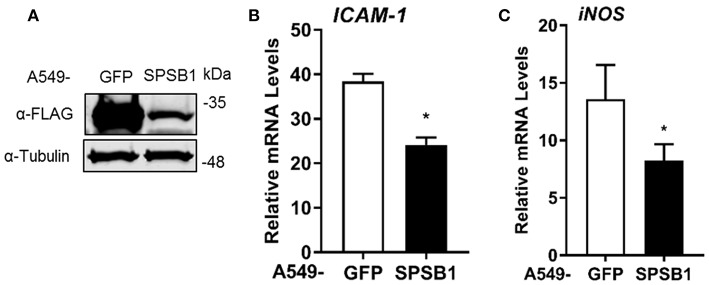
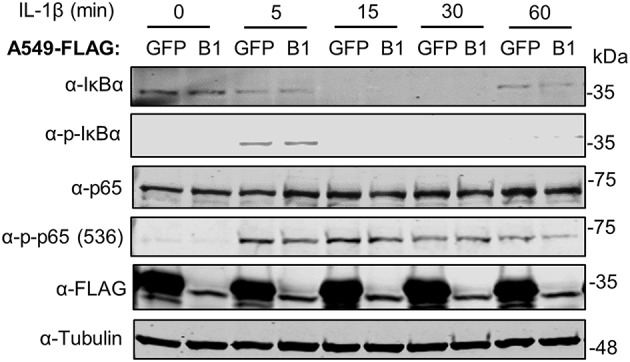
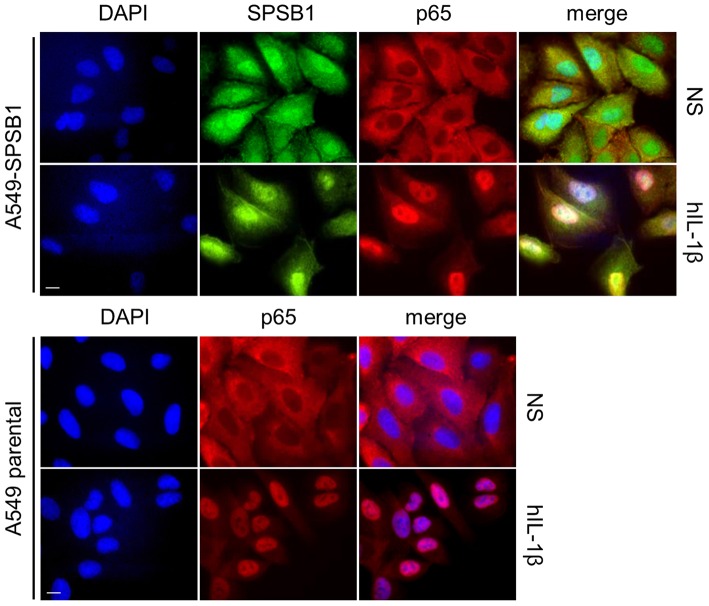
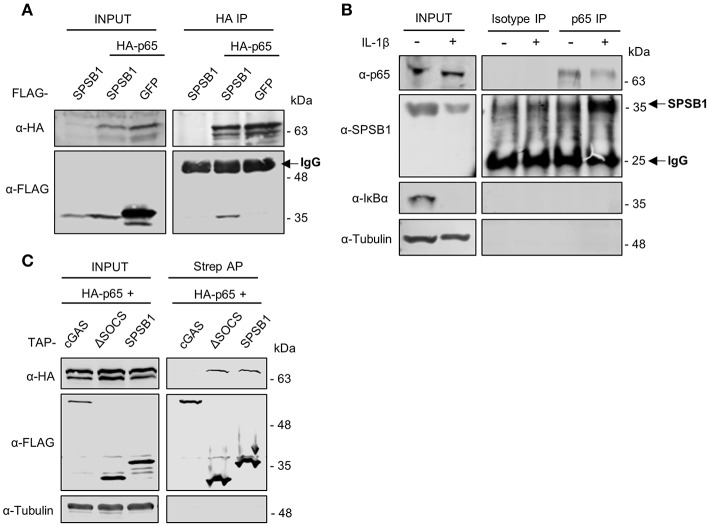
The initiation of innate immune responses against pathogens relies on the activation of pattern-recognition receptors (PRRs) and corresponding intracellular signaling cascades. To avoid inappropriate or excessive activation of PRRs, these responses are tightly controlled. Cullin-RING E3 ubiquitin ligases (CRLs) have emerged as critical regulators of many cellular functions including innate immune activation and inflammation. CRLs form multiprotein complexes in which a Cullin protein acts as a scaffold and recruits specific adaptor proteins, which in turn recognize specific substrate proteins for ubiquitylation, hence providing selectivity. CRLs are divided into 5 main groups, each of which uses a specific group of adaptor proteins. Here, we systematically depleted all predicted substrate adaptors for the CRL5 family (the so-called SOCS-box proteins) and assessed the impact on the activation of the inflammatory transcription factor NF-κB. Depletion of SPSB1 resulted in a significant increase in NF-κB activation, indicating the importance of SPSB1 as an NF-κB negative regulator. In agreement, overexpression of SPSB1 suppressed NF-κB activity in a potent, dose-dependent manner in response to various agonists. Inhibition by SPSB1 was specific to NF-κB, because other transcription factors related to innate immunity and interferon (IFN) responses such as IRF-3, AP-1, and STATs remained unaffected by SPSB1. SPSB1 suppressed NF-κB activation induced via multiple pathways including Toll-like receptors and RNA and DNA sensing adaptors, and required the presence of its SOCS-box domain. To provide mechanistic insight, we examined phosphorylation and degradation of the inhibitor of κB (IκBα) and p65 translocation into the nucleus. Both remained unaffected by SPSB1, indicating that SPSB1 exerts its inhibitory activity downstream, or at the level, of the NF-κB heterodimer. In agreement with this, SPSB1 was found to co-precipitate with p65 after over-expression and at endogenous levels. Additionally, A549 cells stably expressing SPSB1 presented lower cytokine levels including type I IFN in response to cytokine stimulation and virus infection. Taken together, our results reveal novel regulatory mechanisms in innate immune signaling and identify the prominent role of SPSB1 in limiting NF-κB activation. Our work thus provides insights into inflammation and inflammatory diseases and new opportunities for the therapeutic targeting of NF-κB transcriptional activity.
先天免疫反应针对病原体的启动依赖于模式识别受体(PRRs)的激活和相应的细胞内信号级联反应。为了避免 PRR 的不适当或过度激活,这些反应受到严格控制。Cullin-RING E3 泛素连接酶(CRLs)已成为许多细胞功能(包括先天免疫激活和炎症)的关键调节剂。CRLs 形成多蛋白复合物,其中 Cullin 蛋白作为支架并募集特定的衔接蛋白,后者反过来识别泛素化的特定底物蛋白,从而提供选择性。CRLs 分为 5 个主要组,每个组都使用特定的衔接蛋白组。在这里,我们系统地耗尽了 CRL5 家族(所谓的 SOCS 盒蛋白)的所有预测底物衔接蛋白,并评估了其对炎症转录因子 NF-κB 激活的影响。SPSB1 的耗竭导致 NF-κB 激活显著增加,表明 SPSB1 作为 NF-κB 负调节剂的重要性。一致地,SPSB1 的过表达以剂量依赖的方式强烈抑制各种激动剂诱导的 NF-κB 活性。SPSB1 的抑制作用是 NF-κB 特异性的,因为与先天免疫和干扰素(IFN)反应相关的其他转录因子,如 IRF-3、AP-1 和 STATs,不受 SPSB1 的影响。SPSB1 抑制通过 Toll 样受体和 RNA 和 DNA 感应衔接子诱导的 NF-κB 激活,并且需要其 SOCS 盒结构域的存在。为了提供机制上的见解,我们检查了 NF-κB 抑制剂(IκBα)的磷酸化和降解以及 p65 向核内的易位。SPSB1 对两者均无影响,表明 SPSB1 在 NF-κB 异二聚体的下游或水平发挥其抑制活性。与这一观点一致的是,在过表达和内源性水平时,发现 SPSB1 与 p65 共沉淀。此外,稳定表达 SPSB1 的 A549 细胞在细胞因子刺激和病毒感染后表现出较低的细胞因子水平,包括 I 型 IFN。总之,我们的研究结果揭示了先天免疫信号中的新调节机制,并确定了 SPSB1 在限制 NF-κB 激活中的突出作用。我们的工作为炎症和炎症性疾病提供了新的见解,并为 NF-κB 转录活性的治疗靶向提供了新的机会。