Liu Xueyan, Li Nan, Liu Sheng, Wang Jun, Zhang Ning, Zheng Xubin, Leung Kwong-Sak, Cheng Lixin
Department of Critical Care Medicine, Shenzhen People's Hospital, The Second Clinical Medicine College of Jinan University, Shenzhen, China.
Department of Stomatology Center, Shenzhen People's Hospital, Second Clinical Medicine College of Jinan University, Shenzhen, China.
Front Bioeng Biotechnol. 2019 Nov 26;7:358. doi: 10.3389/fbioe.2019.00358. eCollection 2019.
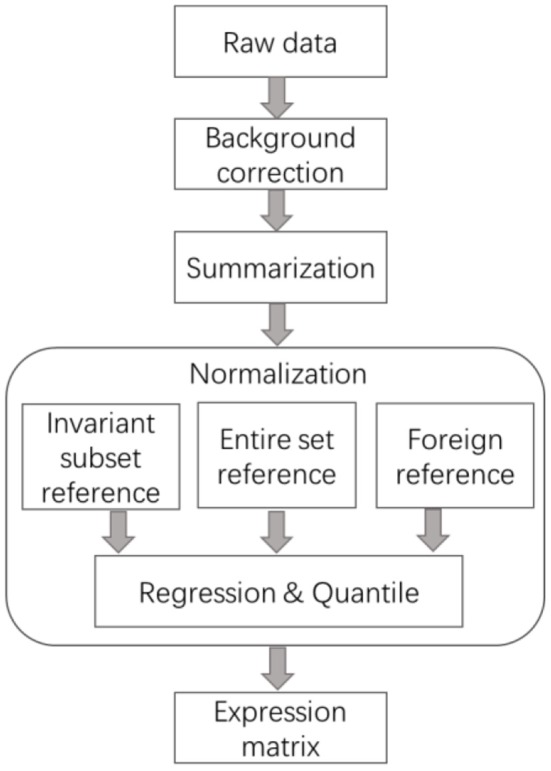
Dozens of normalization methods for correcting experimental variation and bias in high-throughput expression data have been developed during the last two decades. Up to 23 methods among them consider the skewness of expression data between sample states, which are even more than the conventional methods, such as loess and quantile. From the perspective of reference selection, we classified the normalization methods for skewed expression data into three categories, data-driven reference, foreign reference, and entire gene set. We separately introduced and summarized these normalization methods designed for gene expression data with global shift between compared conditions, including both microarray and RNA-seq, based on the reference selection strategies. To our best knowledge, this is the most comprehensive review of available preprocessing algorithms for the unbalanced transcriptome data. The anatomy and summarization of these methods shed light on the understanding and appropriate application of preprocessing methods.
在过去二十年中,已经开发出了数十种用于校正高通量表达数据中的实验变异和偏差的标准化方法。其中多达23种方法考虑了样本状态之间表达数据的偏度,这甚至比传统方法(如局部加权回归和分位数法)还要多。从参考选择的角度来看,我们将针对偏态表达数据的标准化方法分为三类:数据驱动参考、外部参考和全基因集。我们基于参考选择策略,分别介绍并总结了为比较条件之间存在全局偏移的基因表达数据(包括微阵列和RNA测序数据)设计的这些标准化方法。据我们所知,这是对可用的非平衡转录组数据预处理算法最全面的综述。这些方法的剖析和总结有助于深入理解和正确应用预处理方法。