Olayemi Ayodeji, Adesina Adetunji Samuel, Strecker Thomas, Magassouba N'Faly, Fichet-Calvet Elisabeth
Natural History Museum, Obafemi Awolowo University, Ile Ife HO220005, Nigeria.
Department of Biochemistry and Molecular Biology, Obafemi Awolowo University, Ile Ife HO220005, Nigeria.
Biology (Basel). 2020 Feb 7;9(2):26. doi: 10.3390/biology9020026.
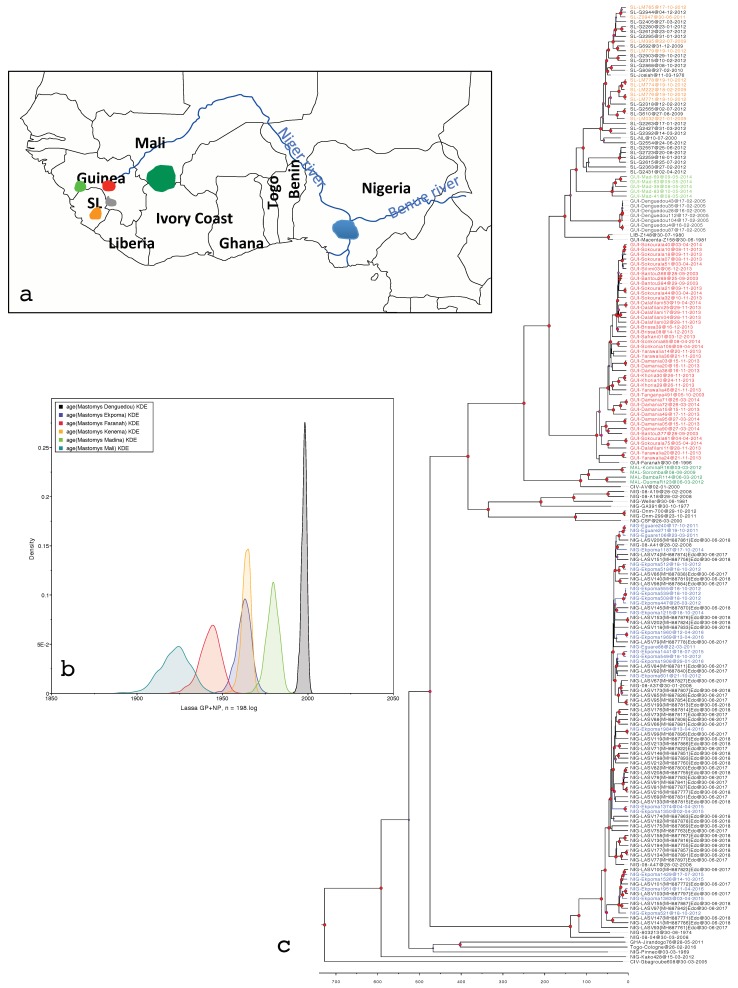
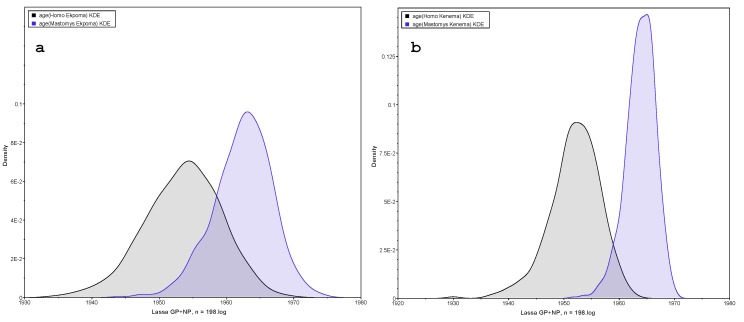
Lassa fever is a viral hemorrhagic illness responsible for thousands of human deaths in West Africa yearly. Rodents are known as natural reservoirs of the causative Lassa mammarenavirus (LASV) while humans are regarded as incidental, spill-over hosts. Analysis of genetic sequences continues to add to our understanding of the evolutionary history, emergence patterns, and the epidemiology of LASV. Hitherto, the source of data in such investigations has mainly comprised human clinical samples. Presently, a rise in the quantity of virus strains accessed through ecological studies over the last 15 years now allows us to explore how LASV sequences obtained from rodents might affect phylogenetic patterns. In this study, we phylogenetically compared LASV sequences obtained from both rodents and humans across West Africa, including those from two localities highly endemic for the disease: Ekpoma in Nigeria and Kenema in Sierra Leone. We performed a time-calibrated phylogeny, using a Bayesian analysis on 198 taxa, including 102 sequences from rodents and 96 from humans. Contrary to expectation, our results show that LASV strains detected in humans within these localities, even those sampled recently, are consistently ancient to those circulating in rodents in the same area. We discuss the possibilities connected to this preliminary outcome. We also propose modalities to guide more comprehensive comparisons of human and rodent data in LASV molecular epidemiological studies.
拉沙热是一种病毒性出血热疾病,每年在西非导致数千人死亡。啮齿动物是致病性拉沙哺乳病毒(LASV)的天然宿主,而人类被视为偶然的溢出宿主。对基因序列的分析不断加深我们对LASV进化史、出现模式和流行病学的理解。迄今为止,此类研究中的数据来源主要包括人类临床样本。目前,过去15年通过生态学研究获得的病毒株数量有所增加,这使我们能够探索从啮齿动物获得的LASV序列如何影响系统发育模式。在这项研究中,我们对从西非啮齿动物和人类中获得的LASV序列进行了系统发育比较,包括来自该病高度流行的两个地区的序列:尼日利亚的埃克波马和塞拉利昂的凯内马。我们使用贝叶斯分析对198个分类单元进行了时间校准的系统发育分析,其中包括102个来自啮齿动物的序列和96个来自人类的序列。与预期相反,我们的结果表明,在这些地区的人类中检测到的LASV毒株,即使是最近采样的毒株,在同一地区的啮齿动物中传播的毒株相比,始终更为古老。我们讨论了与这一初步结果相关的可能性。我们还提出了一些方法,以指导在LASV分子流行病学研究中对人类和啮齿动物数据进行更全面的比较。