Department of Pathophysiology, The Key Immunopathology Laboratory of Guangdong Province, Shantou University Medical College, Guangdong 515031, China.
Department of physiology, Shantou University Medical College, Guangdong 515031, China.
Biomolecules. 2020 Feb 10;10(2):266. doi: 10.3390/biom10020266.
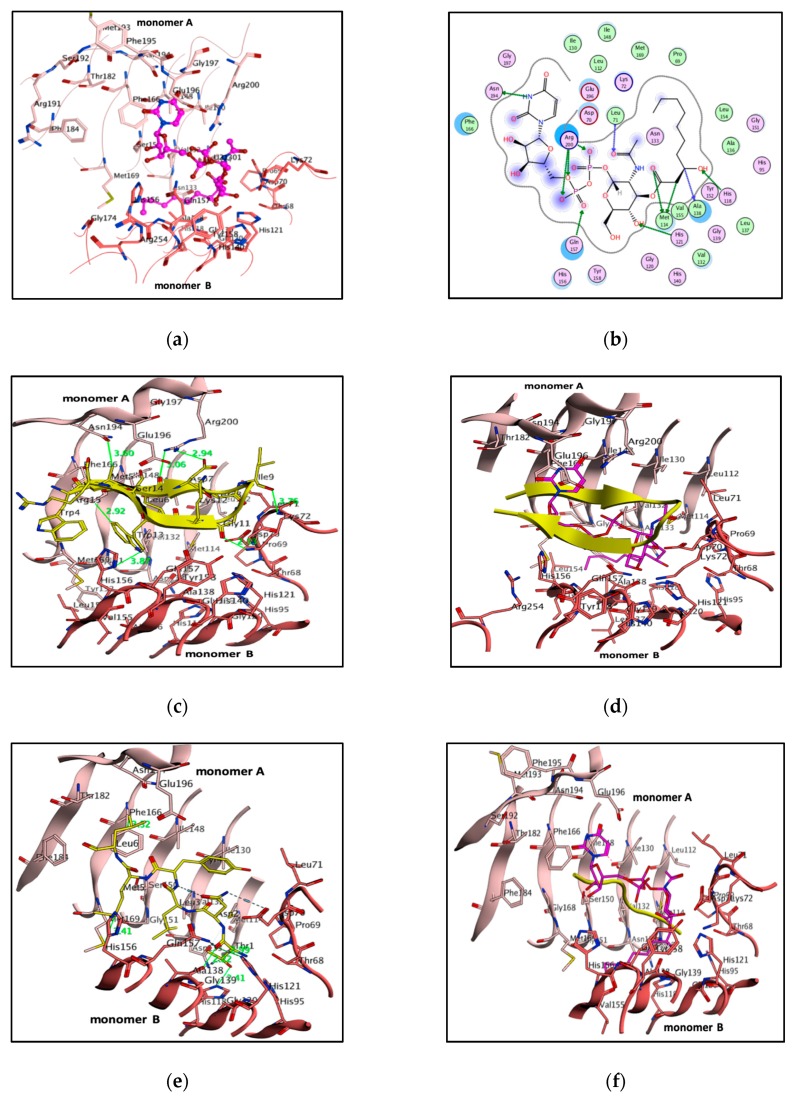
Multidrug resistance in Pseudomonas aeruginosa is a noticeable and ongoing major obstacle for inhibitor design. In uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) acetyltransferase (PaLpxA) is an essential enzyme of lipid A biosynthesis and an attractive drug target. PaLpxA is a homotrimer, and the binding pocket for its substrate, UDP-GlcNAc, is positioned between the monomer A-monomer B interface. The uracil moiety binds at one monomer A, the GlcNAc moiety binds at another monomer B, and a diphosphate form bonds with both monomers. The catalytic residues are conserved and display a similar catalytic mechanism across orthologs, but some distinctions exist between pocket sizes, residue differences, substrate positioning and specificity. The analysis of diversified pockets, volumes, and ligand positions was determined between orthologues that could aid in selective inhibitor development. Thenceforth, a complex-based pharmacophore model was generated and subjected to virtual screening to identify compounds with similar pharmacophoric properties. Docking and general Born-volume integral (GBVI) studies demonstrated 10 best lead compounds with selective inhibition properties with essential residues in the pocket. For biological access, these scaffolds complied with the Lipinski rule, no toxicity and drug likeness properties, and were considered as lead compounds. Hence, these scaffolds could be helpful for the development of potential selective PaLpxA inhibitors.
铜绿假单胞菌的多药耐药性是抑制剂设计中一个显著且持续存在的主要障碍。在尿苷二磷酸 N-乙酰葡萄糖胺 (UDP-GlcNAc) 乙酰基转移酶 (PaLpxA) 是脂质 A 生物合成的必需酶,也是一个有吸引力的药物靶点。PaLpxA 是一个同源三聚体,其底物 UDP-GlcNAc 的结合口袋位于单体 A-单体 B 界面之间。尿嘧啶部分结合在一个单体 A 上,GlcNAc 部分结合在另一个单体 B 上,二磷酸形式与两个单体结合。催化残基在同源物中保守,显示出相似的催化机制,但口袋大小、残基差异、底物定位和特异性之间存在一些区别。对不同口袋、体积和配体位置的分析是在同源物之间进行的,这有助于选择性抑制剂的开发。此后,生成了基于复合物的药效团模型,并进行虚拟筛选,以鉴定具有相似药效团特性的化合物。对接和一般 Born 体积积分 (GBVI) 研究表明,口袋中具有必需残基的 10 个最佳先导化合物具有选择性抑制特性。为了生物利用度,这些支架符合 Lipinski 规则,没有毒性和药物相似性特性,被认为是先导化合物。因此,这些支架可能有助于开发潜在的选择性 PaLpxA 抑制剂。