MIND Institute and Department of Psychiatry and Behavioral Sciences, University of California Davis School of Medicine, Sacramento, CA, USA.
Mouse Imaging Centre, Toronto Centre for Phenogenomics, The Hospital for Sick Children, Toronto, ON, Canada.
Transl Psychiatry. 2020 Jan 27;10(1):39. doi: 10.1038/s41398-020-0720-2.
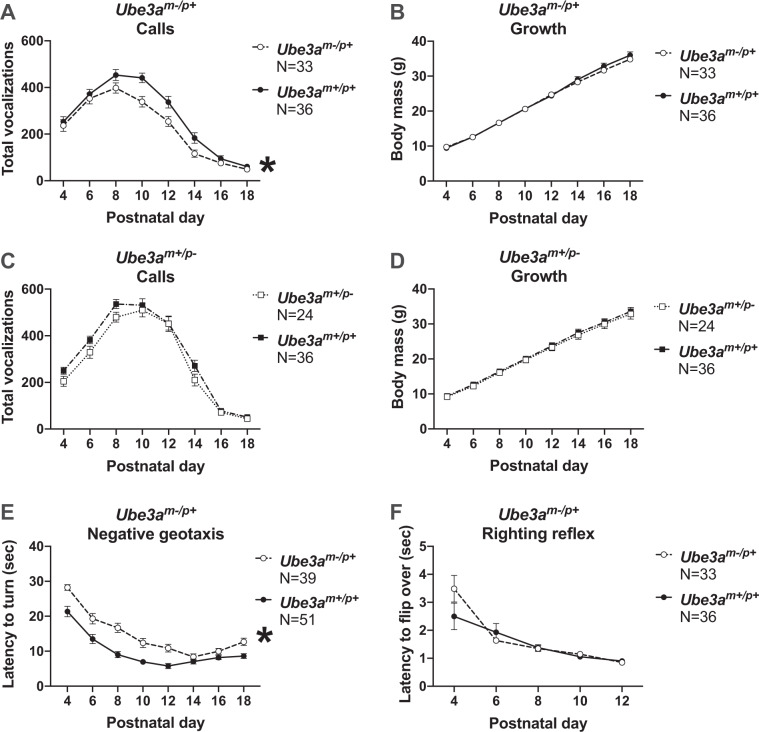
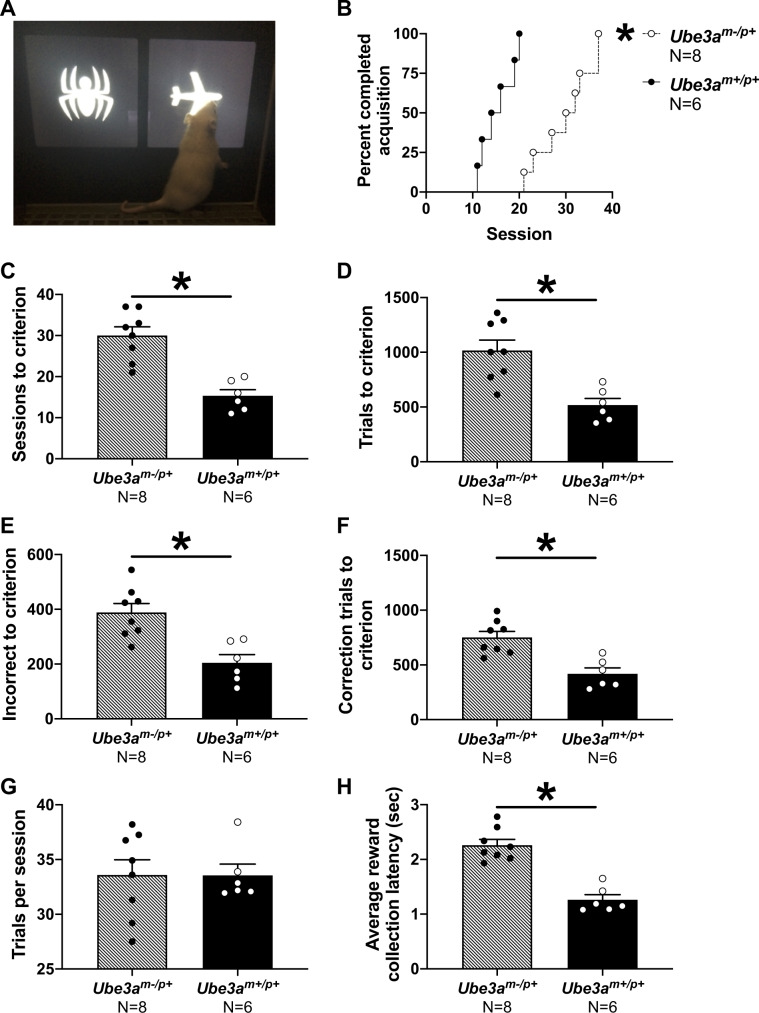
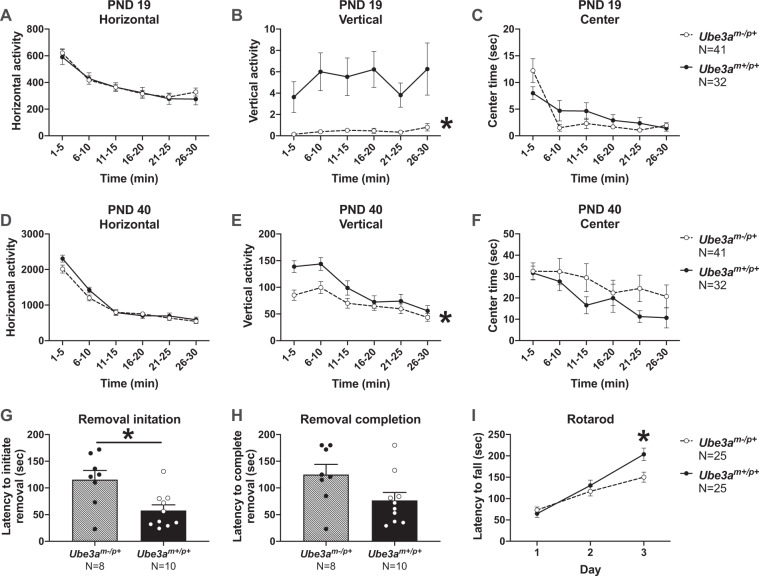
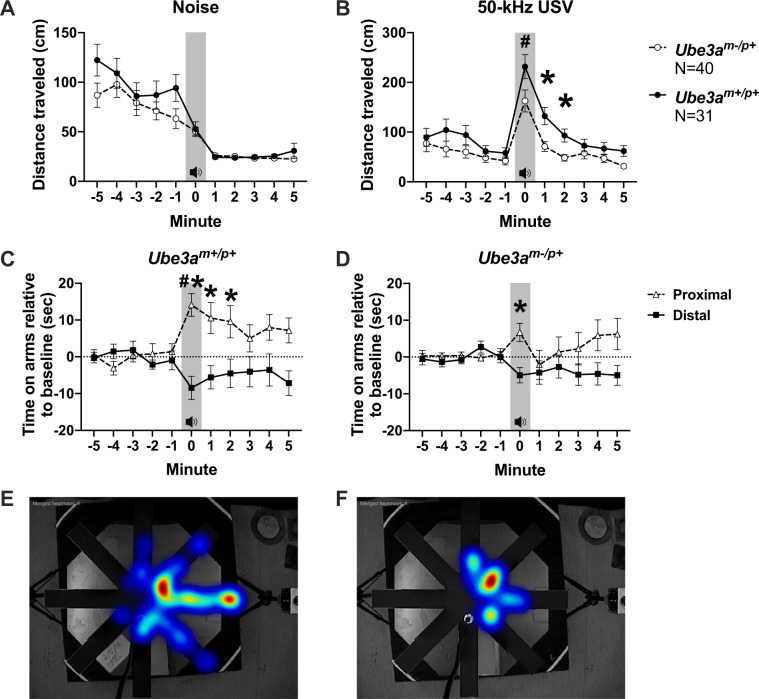
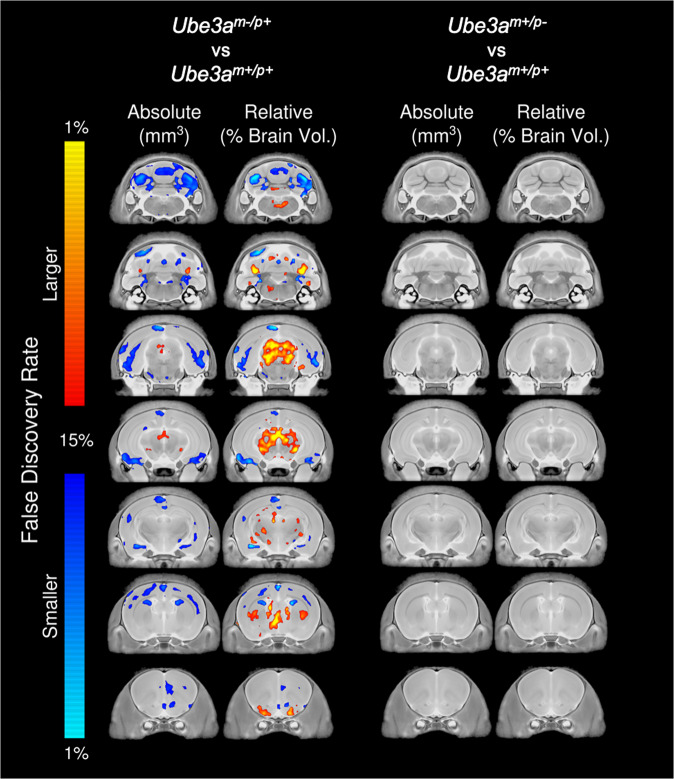
Angelman syndrome (AS) is a rare neurodevelopmental disorder characterized by developmental delay, impaired communication, motor deficits and ataxia, intellectual disabilities, microcephaly, and seizures. The genetic cause of AS is the loss of expression of UBE3A (ubiquitin protein ligase E6-AP) in the brain, typically due to a deletion of the maternal 15q11-q13 region. Previous studies have been performed using a mouse model with a deletion of a single exon of Ube3a. Since three splice variants of Ube3a exist, this has led to a lack of consistent reports and the theory that perhaps not all mouse studies were assessing the effects of an absence of all functional UBE3A. Herein, we report the generation and functional characterization of a novel model of Angelman syndrome by deleting the entire Ube3a gene in the rat. We validated that this resulted in the first comprehensive gene deletion rodent model. Ultrasonic vocalizations from newborn Ube3a were reduced in the maternal inherited deletion group with no observable change in the Ube3a paternal transmission cohort. We also discovered Ube3a exhibited delayed reflex development, motor deficits in rearing and fine motor skills, aberrant social communication, and impaired touchscreen learning and memory in young adults. These behavioral deficits were large in effect size and easily apparent in the larger rodent species. Low social communication was detected using a playback task that is unique to rats. Structural imaging illustrated decreased brain volume in Ube3a and a variety of intriguing neuroanatomical phenotypes while Ube3a did not exhibit altered neuroanatomy. Our report identifies, for the first time, unique AS relevant functional phenotypes and anatomical markers as preclinical outcomes to test various strategies for gene and molecular therapies in AS.
天使综合征(AS)是一种罕见的神经发育障碍,其特征是发育迟缓、沟通障碍、运动缺陷和共济失调、智力障碍、小头畸形和癫痫发作。AS 的遗传原因是大脑中 UBE3A(泛素蛋白连接酶 E6-AP)表达的丧失,通常是由于母系 15q11-q13 区域的缺失。以前的研究使用的是 Ube3a 单一外显子缺失的小鼠模型。由于 Ube3a 存在三种剪接变体,这导致了缺乏一致的报告,以及并非所有小鼠研究都在评估缺乏所有功能性 UBE3A 的影响的理论。在此,我们报告了通过删除大鼠 Ube3a 基因的全部来生成和功能表征新型天使综合征模型。我们验证了这导致了第一个全面的基因缺失啮齿动物模型。在母系遗传缺失组中,新生 Ube3a 的超声发声减少,而在 Ube3a 父系传递组中没有观察到可观察到的变化。我们还发现 Ube3a 表现出反射发育延迟、饲养和精细运动技能运动缺陷、异常社会交流以及年轻成年人的触摸屏学习和记忆受损。这些行为缺陷的效应量很大,在较大的啮齿动物物种中很容易观察到。使用独特的大鼠播放任务检测到低社会交流。结构成像说明了 Ube3a 和各种有趣的神经解剖表型的脑体积减少,而 Ube3a 没有表现出改变的神经解剖结构。我们的报告首次确定了独特的 AS 相关功能表型和解剖学标记作为临床前结果,以测试各种 AS 基因和分子治疗策略。