Instituto de Telecomunicações, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, Lisboa, 1049-001, Portugal.
INESC-ID, Instituto Superior Técnico, Universidade de Lisboa, Rua Alves Redol 9, Lisboa, 1000-029, Portugal.
BMC Bioinformatics. 2020 Feb 18;21(1):59. doi: 10.1186/s12859-020-3390-4.
Understanding cellular and molecular heterogeneity in glioblastoma (GBM), the most common and aggressive primary brain malignancy, is a crucial step towards the development of effective therapies. Besides the inter-patient variability, the presence of multiple cell populations within tumors calls for the need to develop modeling strategies able to extract the molecular signatures driving tumor evolution and treatment failure. With the advances in single-cell RNA Sequencing (scRNA-Seq), tumors can now be dissected at the cell level, unveiling information from their life history to their clinical implications.
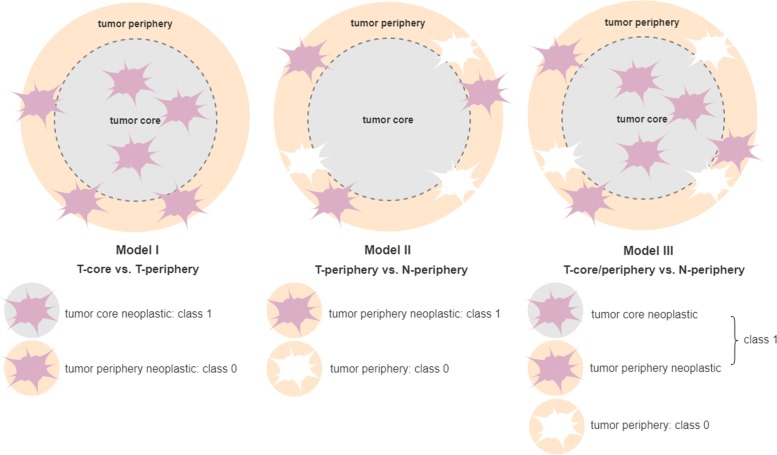
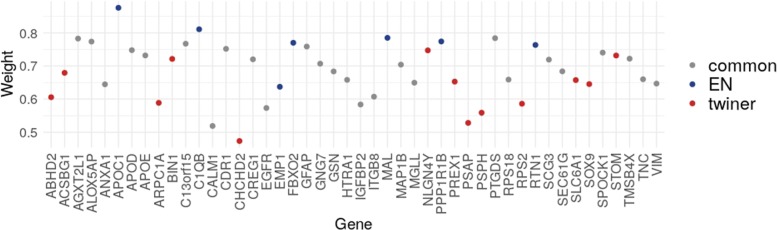
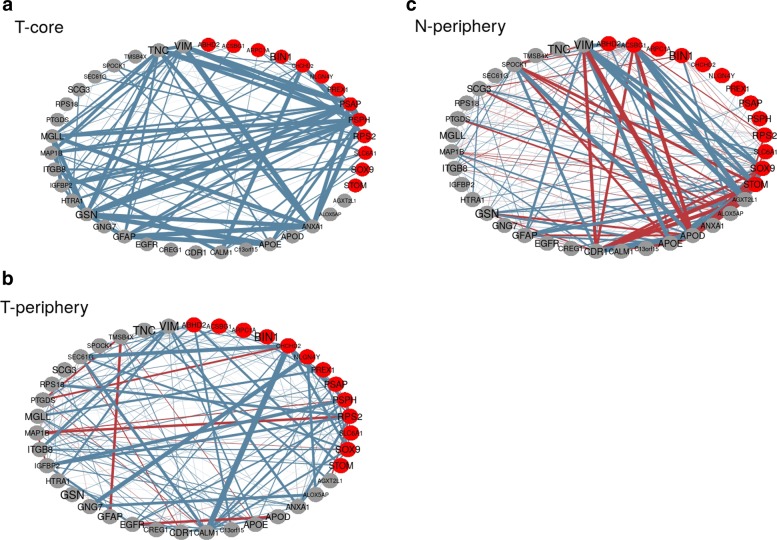
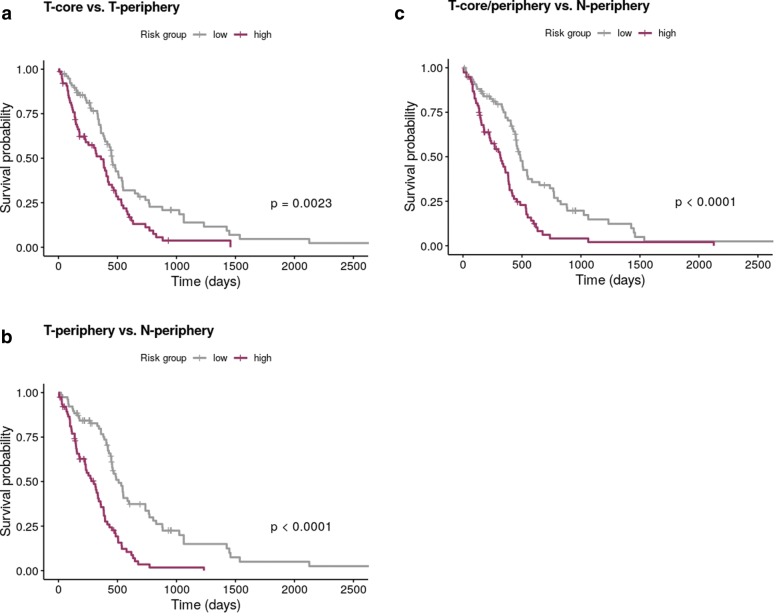
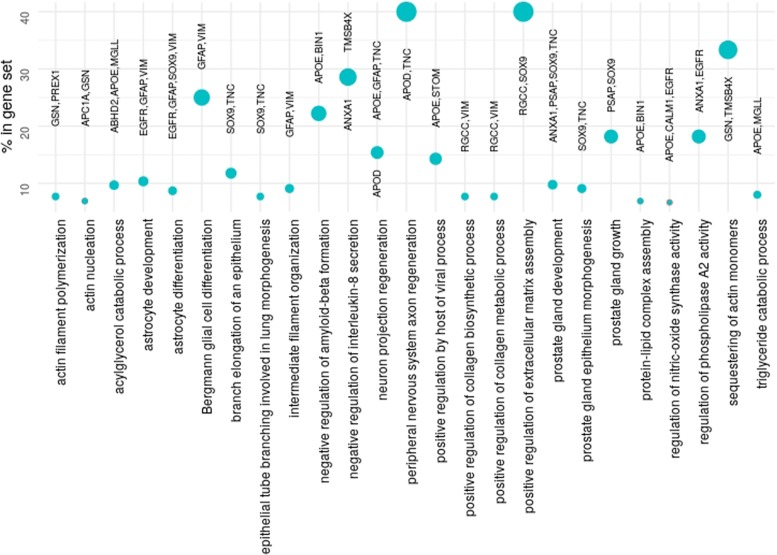
We propose a classification setting based on GBM scRNA-Seq data, through sparse logistic regression, where different cell populations (neoplastic and normal cells) are taken as classes. The goal is to identify gene features discriminating between the classes, but also those shared by different neoplastic clones. The latter will be approached via the network-based twiner regularizer to identify gene signatures shared by neoplastic cells from the tumor core and infiltrating neoplastic cells originated from the tumor periphery, as putative disease biomarkers to target multiple neoplastic clones. Our analysis is supported by the literature through the identification of several known molecular players in GBM. Moreover, the relevance of the selected genes was confirmed by their significance in the survival outcomes in bulk GBM RNA-Seq data, as well as their association with several Gene Ontology (GO) biological process terms.
We presented a methodology intended to identify genes discriminating between GBM clones, but also those playing a similar role in different GBM neoplastic clones (including migrating cells), therefore potential targets for therapy research. Our results contribute to a deeper understanding on the genetic features behind GBM, by disclosing novel therapeutic directions accounting for GBM heterogeneity.
了解胶质母细胞瘤(GBM)——最常见和侵袭性最强的原发性脑恶性肿瘤——的细胞和分子异质性,是开发有效治疗方法的关键步骤。除了患者间的变异性外,肿瘤内存在多个细胞群体也需要开发能够提取驱动肿瘤进化和治疗失败的分子特征的建模策略。随着单细胞 RNA 测序(scRNA-Seq)的进展,现在可以在细胞水平上对肿瘤进行解剖,揭示其从生命史到临床意义的信息。
我们通过稀疏逻辑回归提出了一种基于 GBM scRNA-Seq 数据的分类设置,其中不同的细胞群体(肿瘤和正常细胞)被视为类别。目标是识别区分类别的基因特征,以及不同肿瘤克隆共有的基因特征。后者将通过基于网络的 twiner 正则化器来识别肿瘤核心和源自肿瘤外围的浸润性肿瘤细胞中的肿瘤细胞共有的基因特征,作为针对多个肿瘤克隆的潜在疾病生物标志物。通过鉴定 GBM 中的几个已知分子参与者,我们的分析得到了文献的支持。此外,通过在批量 GBM RNA-Seq 数据中的生存结果的显著性以及与几个基因本体(GO)生物学过程术语的相关性,验证了所选基因的相关性。
我们提出了一种旨在识别区分 GBM 克隆的基因的方法,还识别了在不同 GBM 肿瘤克隆(包括迁移细胞)中发挥类似作用的基因,因此是治疗研究的潜在靶点。我们的结果通过揭示考虑 GBM 异质性的新治疗方向,为了解 GBM 的遗传特征做出了贡献。