Department of Pharmaceutical Sciences, Northeastern University, 360 Huntington Avenue, Boston, MA 02115, USA.
Departments of Pediatrics and Physiology & Biophysics, University of California, Irvine, 1001 Health Sciences Road, Irvine Hall, Irvine, CA 92697, USA.
Cell Rep. 2020 Feb 18;30(7):2225-2236.e4. doi: 10.1016/j.celrep.2020.01.025.
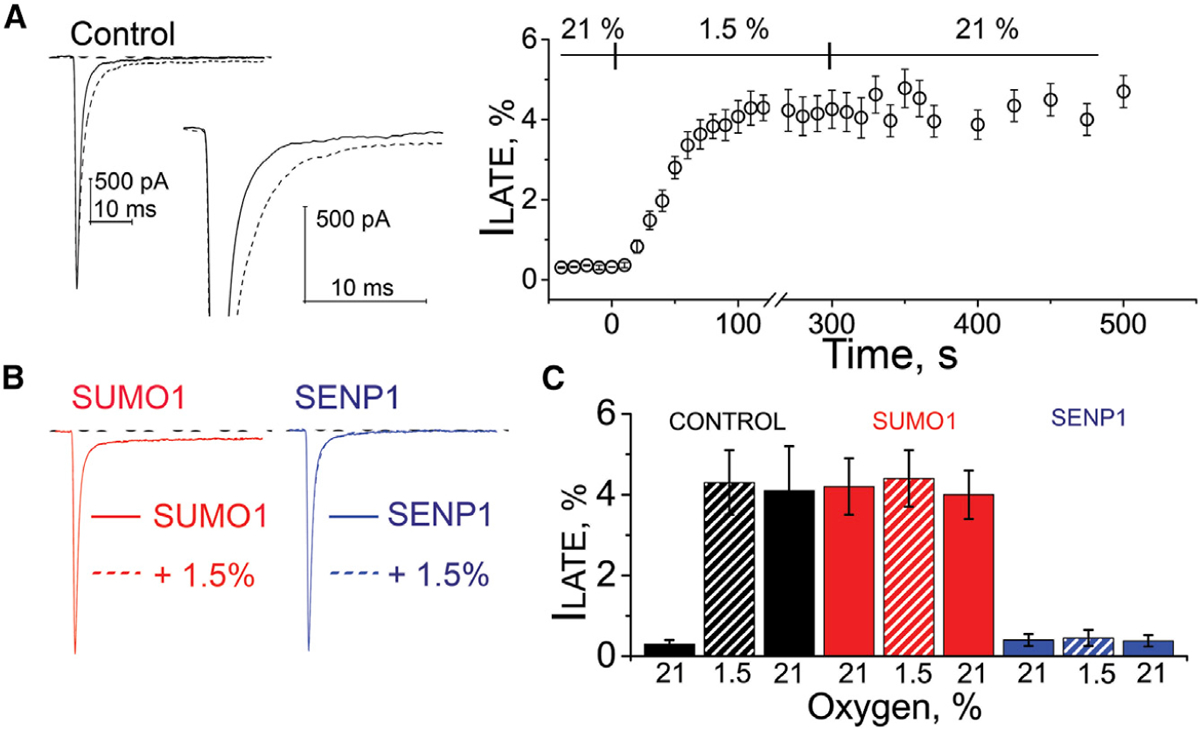
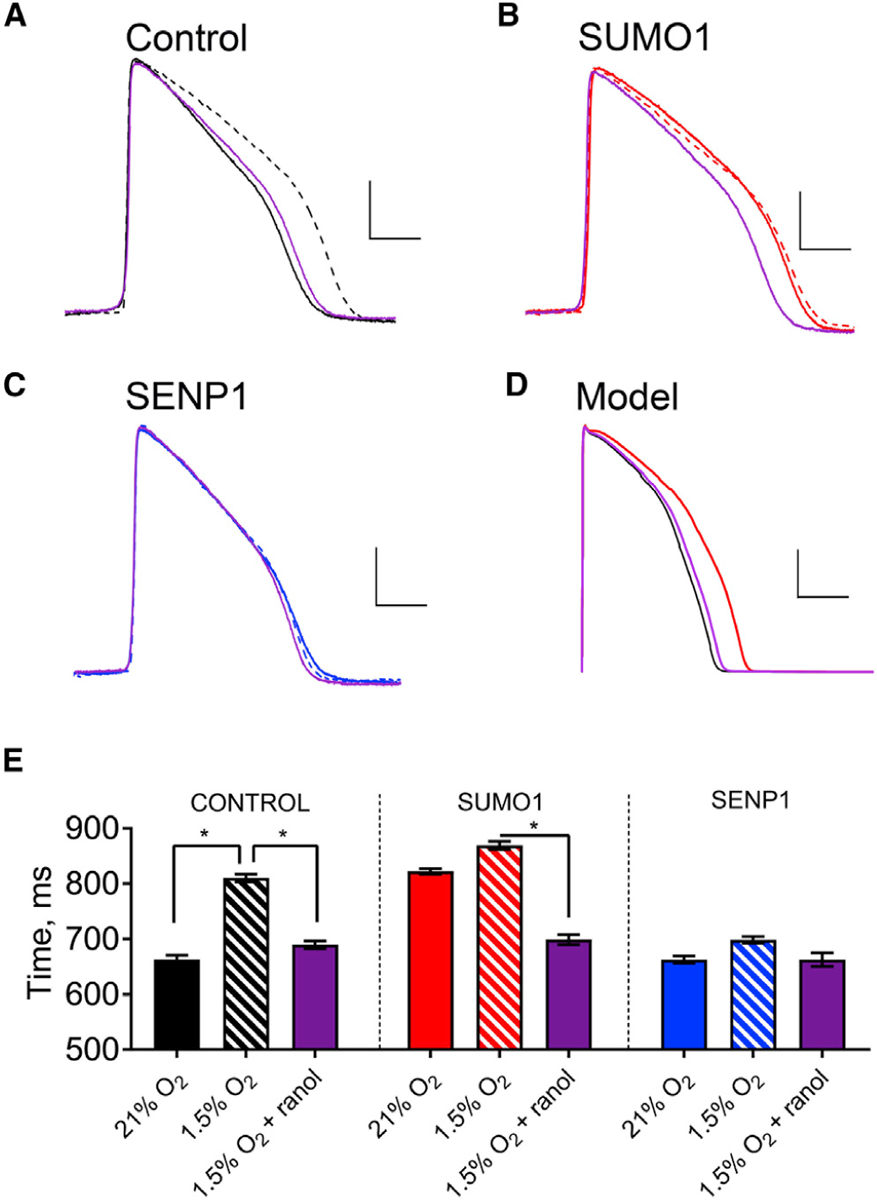
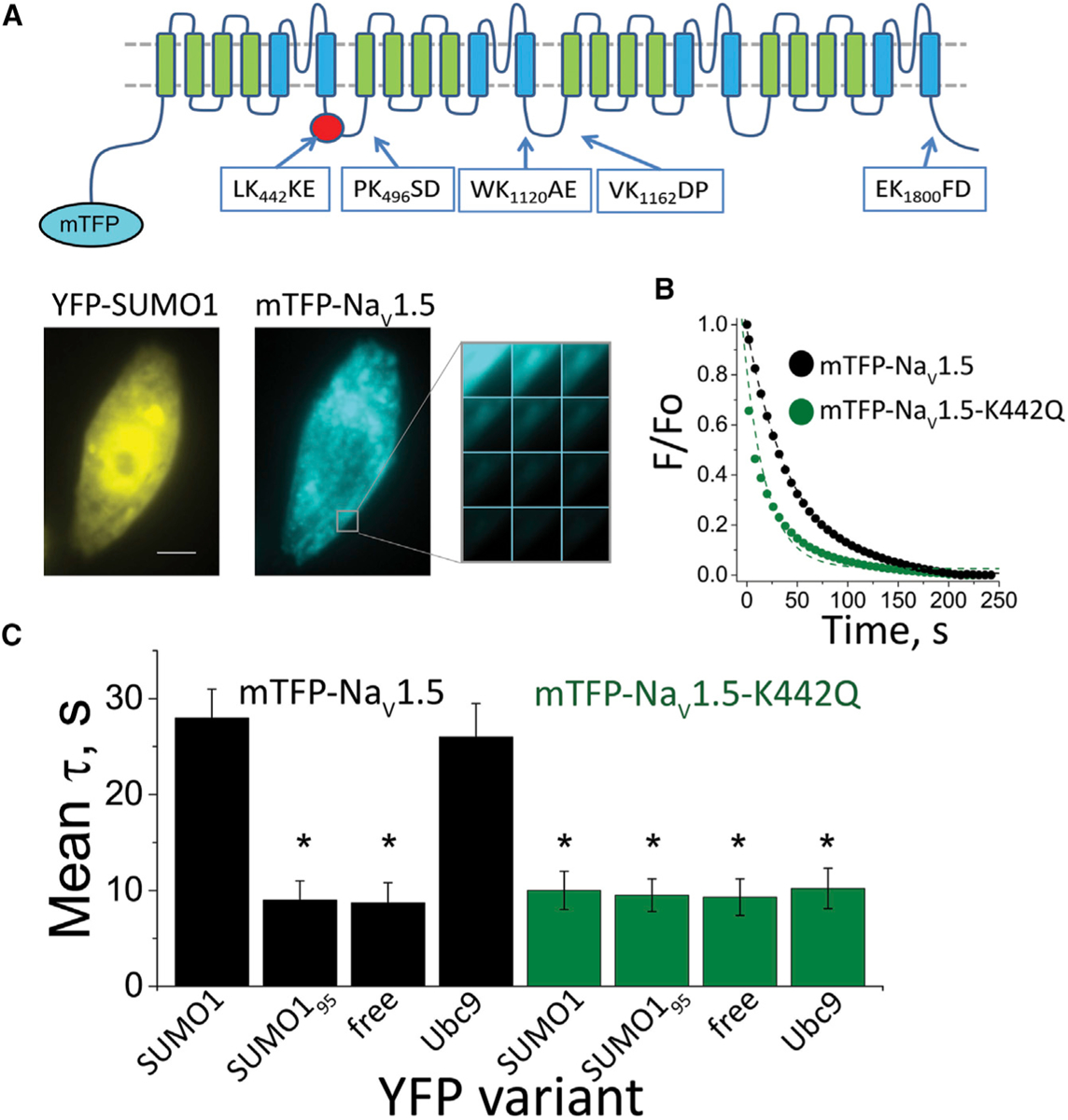
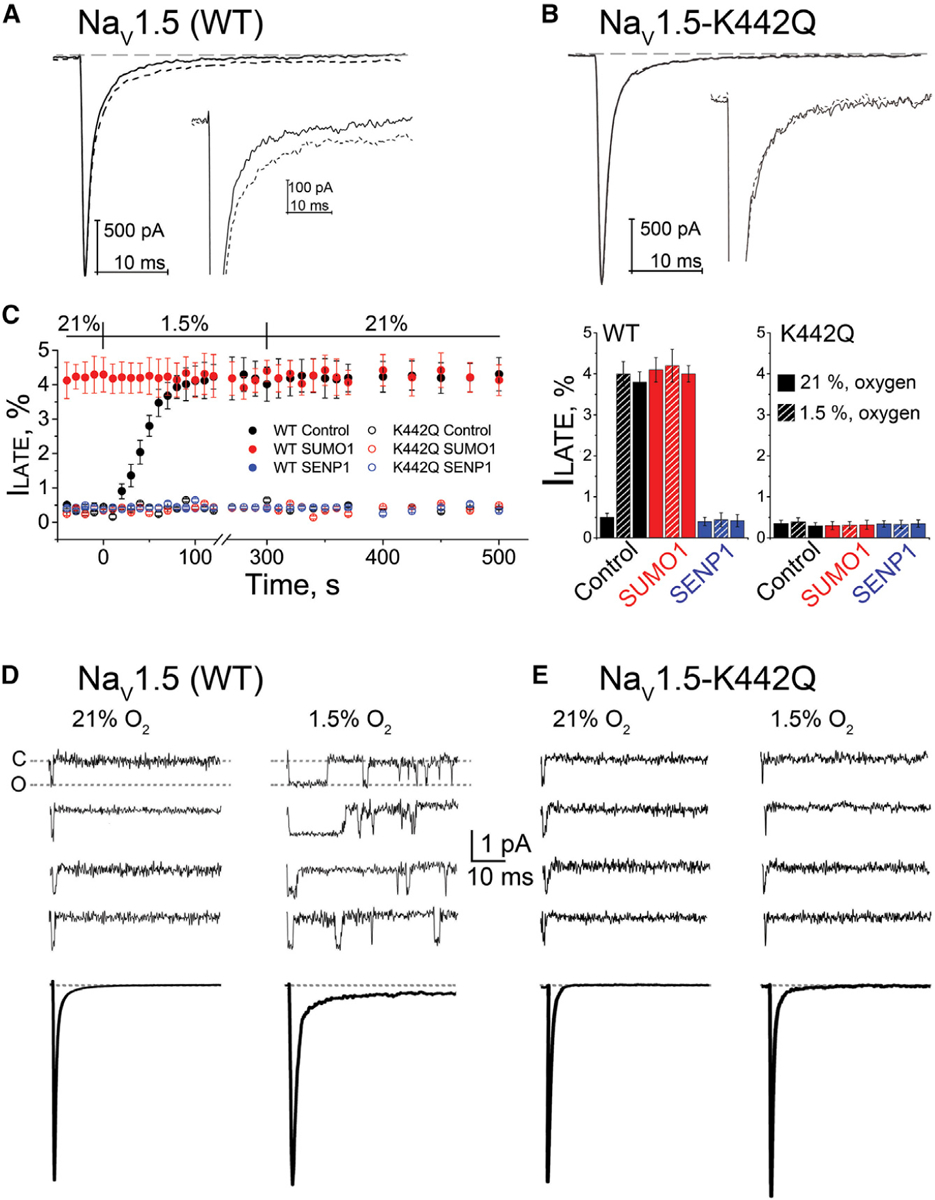
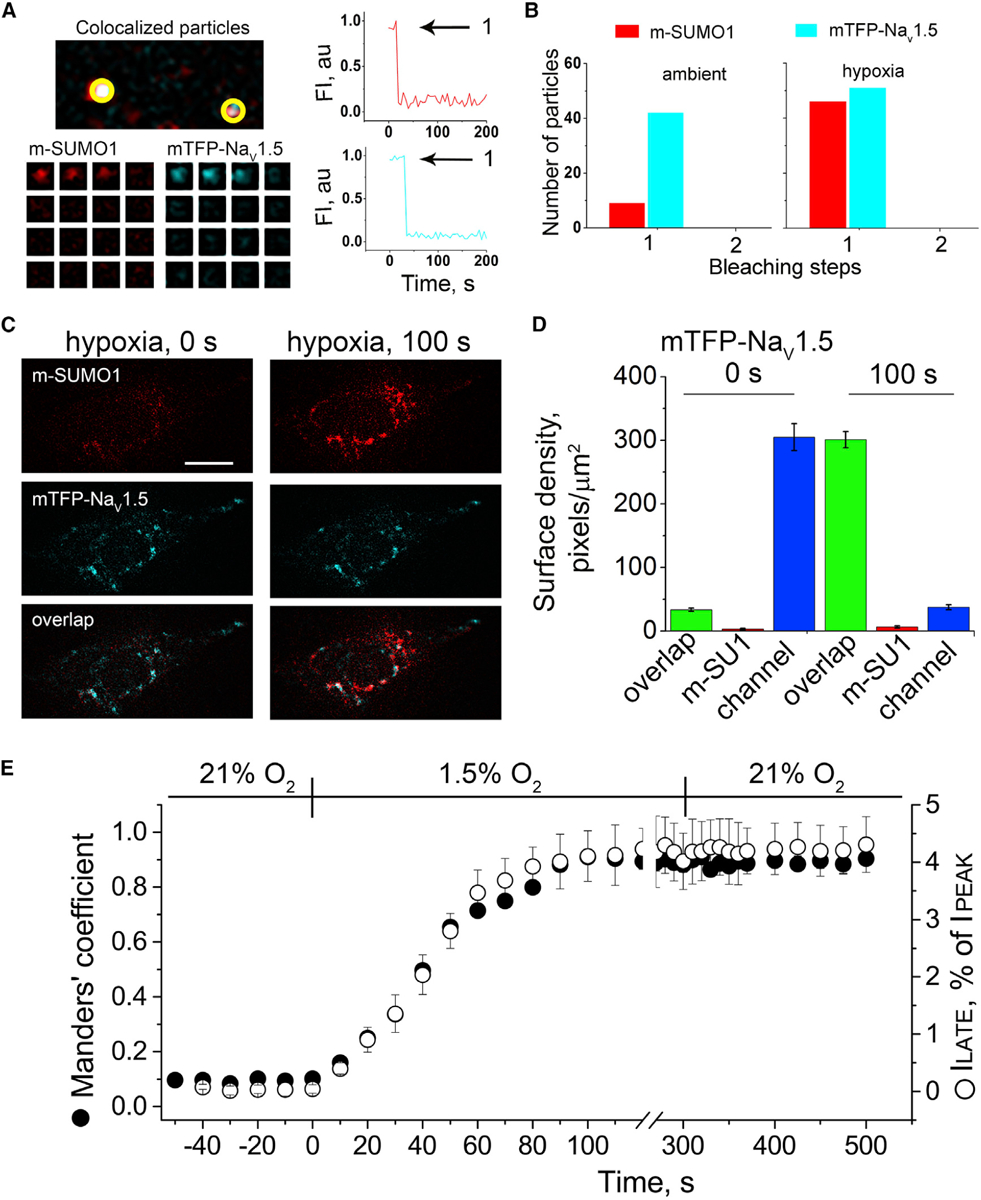
Acute cardiac hypoxia produces life-threatening elevations in late sodium current (I) in the human heart. Here, we show the underlying mechanism: hypoxia induces rapid SUMOylation of Na1.5 channels so they reopen when normally inactive, late in the action potential. Na1.5 is SUMOylated only on lysine 442, and the mutation of that residue, or application of a deSUMOylating enzyme, prevents hypoxic reopenings. The time course of SUMOylation of single channels in response to hypoxia coincides with the increase in I, a reaction that is complete in under 100 s. In human cardiac myocytes derived from pluripotent stem cells, hypoxia-induced I is confirmed to be SUMO-dependent and to produce action potential prolongation, the pro-arrhythmic change observed in patients.
急性心脏缺氧会导致人类心脏晚期钠电流 (I) 产生危及生命的升高。在这里,我们展示了潜在的机制:缺氧诱导 Na1.5 通道的快速 SUMO 化,使它们在动作电位晚期正常失活时重新开放。Na1.5 仅在赖氨酸 442 上 SUMO 化,该残基的突变或 SUMO 去酶的应用可防止缺氧重新开放。对单个通道的 SUMO 化的时间过程与 I 的增加相吻合,该反应在 100 秒内完成。在源自多能干细胞的人心肌细胞中,证实缺氧诱导的 I 是 SUMO 依赖性的,并产生动作电位延长,这是在患者中观察到的致心律失常变化。