Liu Danqi, Zhou Boting, Liu Rangru
Department of Pharmacy, Xiangya Hospital, Central South University, Changsha, People's Republic of China.
Institute for Rational and Safe Medication Practices, National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, People's Republic of China.
PeerJ. 2020 Feb 14;8:e8504. doi: 10.7717/peerj.8504. eCollection 2020.
Gastric carcinoma is a very diverse disease. The progression of gastric carcinoma is influenced by complicated gene networks. This study aims to investigate the actual and potential prognostic biomarkers related to survival in gastric carcinoma patients to further our understanding of tumor biology.
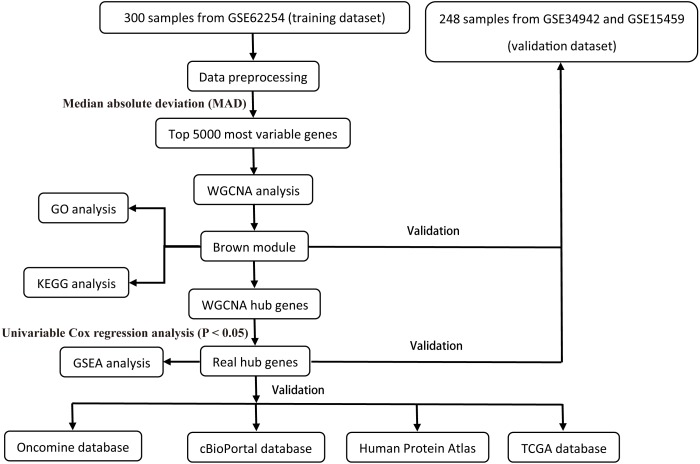
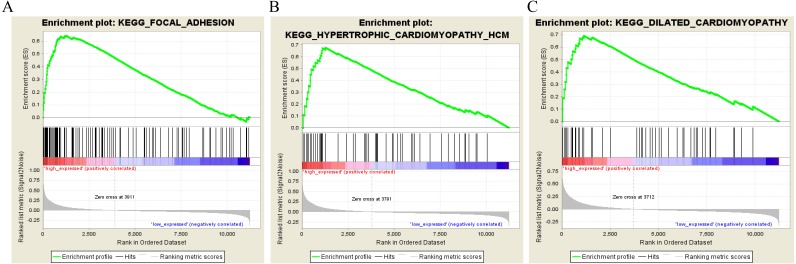
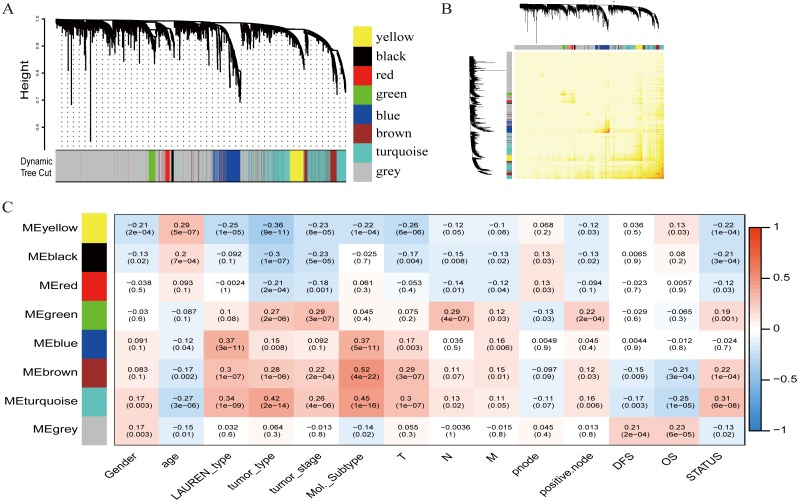
A weighted gene co-expression network analysis was performed with a transcriptome dataset to identify networks and hub genes relevant to gastric carcinoma prognosis. Data was obtained from 300 primary gastric carcinomas (GSE62254). A validation dataset (GSE34942 and GSE15459) and TCGA dataset confirmed the results. Gene ontology, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, and gene set enrichment analysis (GSEA) were performed to identify the clusters responsible for the biological processes and pathways of this disease.
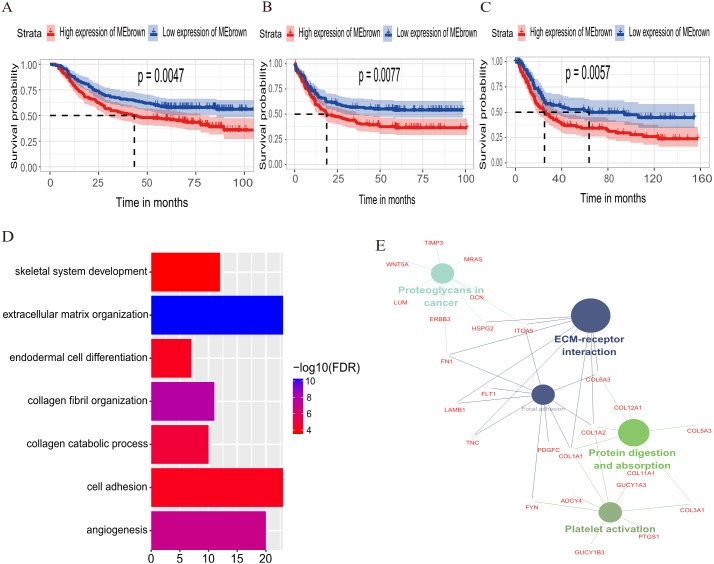
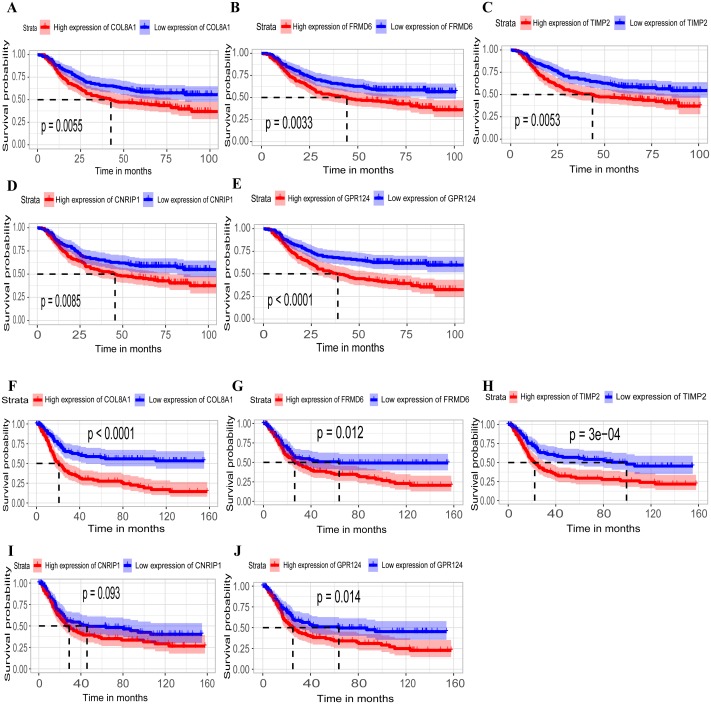
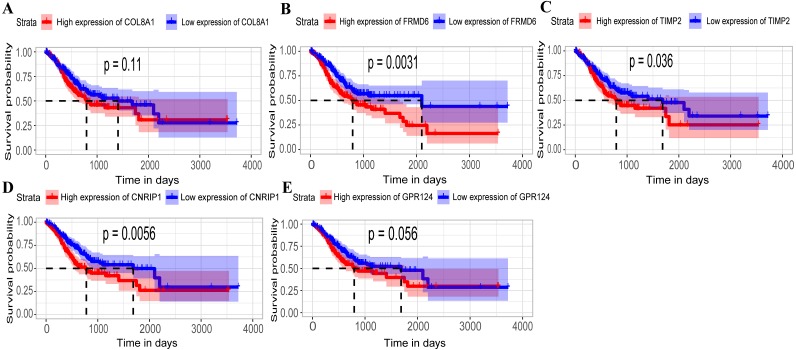
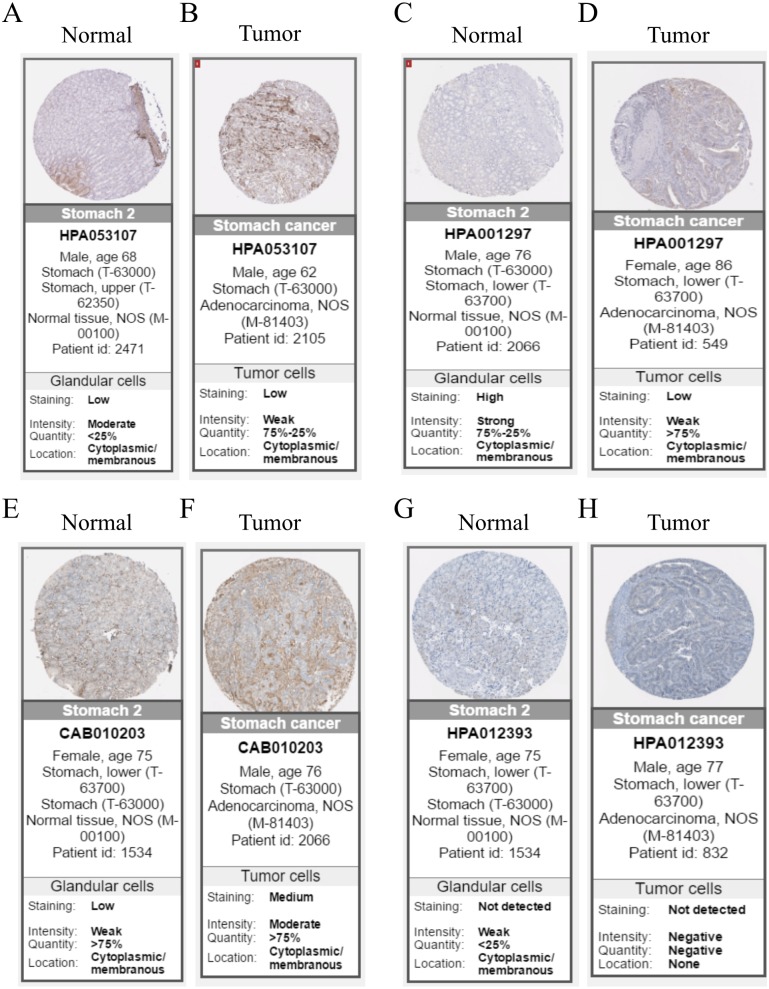
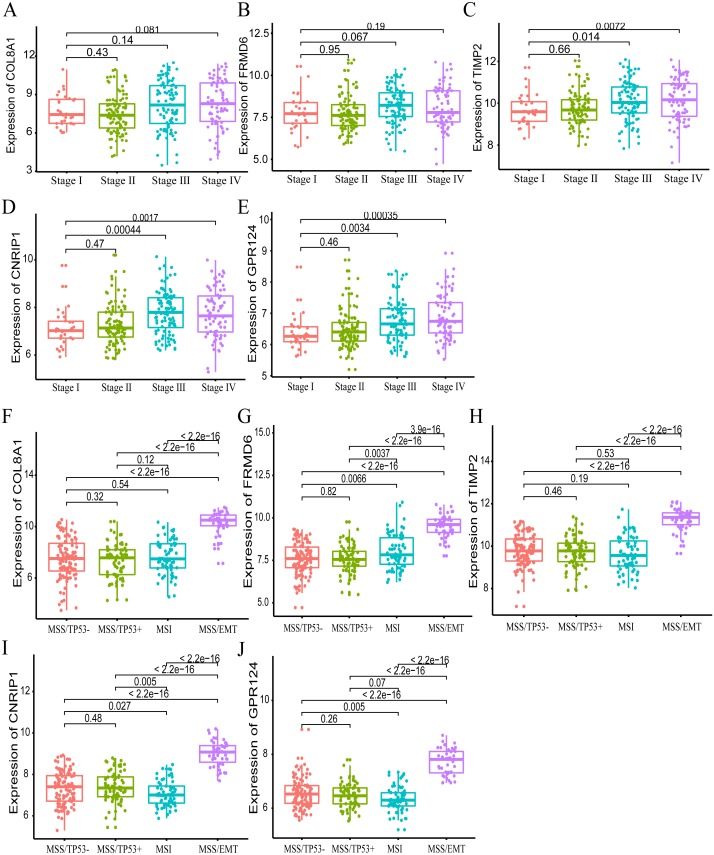
A brown transcriptional module enriched in the organizational process of the extracellular matrix was significantly correlated with overall survival (HR = 1.586, = 0.005, 95% CI [1.149-2.189]) and disease-free survival (HR = 1.544, = 0.008, 95% CI [1.119-2.131]). These observations were confirmed in the validation dataset (HR = 1.664, = 0.006, 95% CI [1.155-2.398] in overall survival). Ten hub genes were identified and confirmed in the validation dataset from this brown module; five key biomarkers (, , , and ) were identified for further research in microsatellite instability (MSI) and epithelial-tomesenchymal transition (MSS/EMT) gastric carcinoma molecular subtypes. A high expression of these genes indicated a poor prognosis.
A transcriptional co-expression network-based approach was used to identify prognostic biomarkers in gastric carcinoma. This method may have potential for use in personalized therapies, however, large-scale randomized controlled clinical trials and replication experiments are needed before these key biomarkers can be applied clinically.
胃癌是一种非常多样化的疾病。胃癌的进展受复杂基因网络的影响。本研究旨在探究与胃癌患者生存相关的实际和潜在预后生物标志物,以加深我们对肿瘤生物学的理解。
利用转录组数据集进行加权基因共表达网络分析,以识别与胃癌预后相关的网络和枢纽基因。数据来自300例原发性胃癌(GSE62254)。一个验证数据集(GSE34942和GSE15459)以及TCGA数据集证实了结果。进行基因本体、京都基因与基因组百科全书(KEGG)通路富集分析以及基因集富集分析(GSEA),以识别负责该疾病生物学过程和通路的聚类。
一个富含细胞外基质组织过程的棕色转录模块与总生存期(HR = 1.586,P = 0.005,95% CI [1.149 - 2.189])和无病生存期(HR = 1.544,P = 0.008,95% CI [1.119 - 2.131])显著相关。这些观察结果在验证数据集中得到证实(总生存期HR = 1.664,P = 0.006,95% CI [1.155 - 2.398])。从这个棕色模块中鉴定出10个枢纽基因,并在验证数据集中得到证实;确定了5个关键生物标志物(、、、和),用于在微卫星不稳定性(MSI)和上皮-间质转化(MSS/EMT)胃癌分子亚型中进一步研究。这些基因的高表达表明预后不良。
采用基于转录共表达网络的方法来识别胃癌的预后生物标志物。该方法可能具有用于个性化治疗的潜力,然而,在这些关键生物标志物能够临床应用之前,需要进行大规模随机对照临床试验和重复实验。