Department of Dermatology, Icahn School of Medicine at Mount Sinai, New York, New York.
Principal Investigator and President, Oregon Medical Research Center, Portland.
JAMA Dermatol. 2020 Apr 1;156(4):411-420. doi: 10.1001/jamadermatol.2020.0079.
Interleukin 13 (IL-13) is a central pathogenic mediator driving multiple features of atopic dermatitis (AD) pathophysiology.
To evaluate the efficacy and safety of lebrikizumab, a novel, high-affinity, monoclonal antibody targeting IL-13 that selectively prevents formation of the IL-13Rα1/IL-4Rα heterodimer receptor signaling complex, in adults with moderate to severe AD.
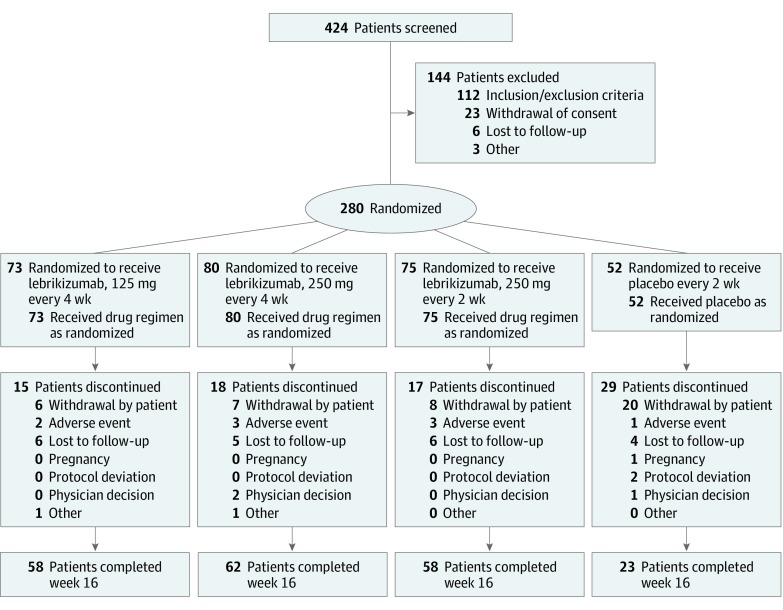
DESIGN, SETTING, AND PARTICIPANTS: A phase 2b, double-blind, placebo-controlled, dose-ranging randomized clinical trial of lebrikizumab injections every 4 weeks or every 2 weeks was conducted from January 23, 2018, to May 23, 2019, at 57 US centers. Participants were adults 18 years or older with moderate to severe AD.
Patients were randomized 2:3:3:3 to placebo every 2 weeks or to subcutaneous injections of lebrikizumab at the following doses: 125 mg every 4 weeks (250-mg loading dose [LD]), 250 mg every 4 weeks (500-mg LD), or 250 mg every 2 weeks (500-mg LD at baseline and week 2).
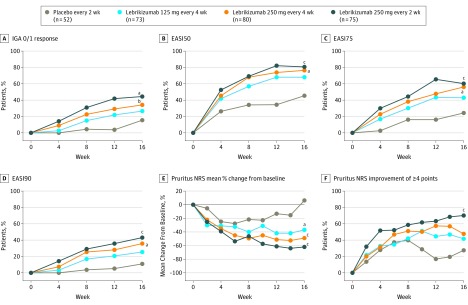
The primary end point was percentage change in the Eczema Area and Severity Index (EASI) (baseline to week 16). Secondary end points for week 16 included proportion of patients achieving Investigator's Global Assessment score of 0 or 1 (IGA 0/1); EASI improvement of at least 50%, 75%, or 90% from baseline; percentage change in the pruritus numeric rating scale (NRS) score; and pruritus NRS score improvement of at least 4 points. Safety assessments included treatment-emergent adverse events.
A total of 280 patients (mean [SD] age, 39.3 [17.5] years; 166 [59.3%] female) were randomized to placebo (n = 52) or to lebrikizumab at doses of 125 mg every 4 weeks (n = 73), 250 mg every 4 weeks (n = 80), or 250 mg every 2 weeks (n = 75). Compared with placebo (EASI least squares mean [SD] percentage change, -41.1% [56.5%]), lebrikizumab groups showed dose-dependent, statistically significant improvement in the primary end point vs placebo at week 16: 125 mg every 4 weeks (-62.3% [37.3%], P = .02), 250 mg every 4 weeks (-69.2% [38.3%], P = .002), and 250 mg every 2 weeks (-72.1% [37.2%], P < .001). Differences vs placebo-treated patients (2 of 44 [4.5%]) in pruritus NRS improvement of at least 4 points were seen as early as day 2 in the high-dose lebrikizumab group (9 of 59 [15.3%]). Treatment-emergent adverse events were reported in 24 of 52 placebo patients (46.2%) and in lebrikizumab patients as follows: 42 of 73 (57.5%) for 125 mg every 4 weeks, 39 of 80 (48.8%) for 250 mg every 4 weeks, and 46 of 75 (61.3%) for 250 mg every 2 weeks; most were mild to moderate and did not lead to discontinuation. Low rates of injection-site reactions (1 of 52 [1.9%] in the placebo group vs 13 of 228 [5.7%] in all lebrikizumab groups), herpesvirus infections (2 [3.8%] vs 8 [3.5%]), and conjunctivitis (0% vs 6 [2.6%]) were reported.
During 16 weeks of treatment, lebrikizumab provided rapid, dose-dependent efficacy across a broad range of clinical manifestations in adult patients with moderate to severe AD and demonstrated a favorable safety profile. These data support the central role of IL-13 in AD pathophysiology. If these findings replicate in phase 3 studies, lebrikizumab may meaningfully advance the standard of care for moderate to severe AD.
ClinicalTrials.gov Identifier: NCT03443024.
重要性:白细胞介素 13(IL-13)是一种主要的致病介质,可驱动特应性皮炎(AD)病理生理学的多种特征。
目的:评估 lebrikizumab 的疗效和安全性,lebrikizumab 是一种新型、高亲和力的单克隆抗体,靶向 IL-13,可选择性地阻止 IL-13Rα1/IL-4Rα 异二聚体受体信号复合物的形成,用于治疗中重度 AD 的成年患者。
设计、地点和参与者:这是一项为期 2b 期、双盲、安慰剂对照、剂量范围的随机临床试验,于 2018 年 1 月 23 日至 2019 年 5 月 23 日在美国 57 个中心进行,共纳入 18 岁及以上的中重度 AD 成年患者。
干预措施:患者按 2:3:3:3 的比例随机分为安慰剂每 2 周组或 lebrikizumab 皮下注射组,剂量分别为每 4 周 125 mg(250-mg 负荷剂量[LD])、每 4 周 250 mg(500-mg LD)或每 2 周 250 mg(基线和第 2 周时为 500-mg LD)。
主要终点和次要终点:主要终点为湿疹面积和严重程度指数(EASI)(基线至第 16 周)的百分比变化。第 16 周的次要终点包括达到研究者全球评估评分 0 或 1(IGA 0/1)的患者比例;EASI 改善至少 50%、75%或 90%;瘙痒数字评分量表(NRS)评分的百分比变化;以及瘙痒 NRS 评分改善至少 4 分。安全性评估包括治疗出现的不良事件。
结果:共有 280 名患者(平均[SD]年龄,39.3[17.5]岁;166[59.3%]为女性)被随机分为安慰剂(n=52)或 lebrikizumab 组,剂量分别为每 4 周 125 mg(n=73)、每 4 周 250 mg(n=80)或每 2 周 250 mg(n=75)。与安慰剂组相比(EASI 最小二乘均值[SD]百分比变化,-41.1%[56.5%]),lebrikizumab 组在第 16 周时显示出剂量依赖性的显著改善:每 4 周 125 mg(-62.3%[37.3%],P=0.02)、每 4 周 250 mg(-69.2%[38.3%],P=0.002)和每 2 周 250 mg(-72.1%[37.2%],P<0.001)。与安慰剂治疗的患者相比(2/44[4.5%]),高剂量 lebrikizumab 组在第 2 天就出现了瘙痒 NRS 评分改善至少 4 分的情况(9/59[15.3%])。在安慰剂组的 52 名患者中,有 24 名(46.2%)和 lebrikizumab 组的患者报告了治疗出现的不良事件,具体为:每 4 周 125 mg 组 42 名(57.5%)、每 4 周 250 mg 组 39 名(48.8%)和每 2 周 250 mg 组 46 名(61.3%);大多数为轻度至中度,并未导致停药。报告了低比例的注射部位反应(安慰剂组 1/52[1.9%],所有 lebrikizumab 组 13/228[5.7%])、疱疹病毒感染(2/52[3.8%],8/228[3.5%])和结膜炎(0%,6/228[2.6%])。
结论和相关性:在 16 周的治疗期间,lebrikizumab 在中重度 AD 成年患者中表现出了快速、剂量依赖性的疗效,跨越了广泛的临床表现,并表现出了良好的安全性特征。这些数据支持白细胞介素 13 在 AD 病理生理学中的核心作用。如果这些发现在 3 期研究中得到复制,lebrikizumab 可能会为中重度 AD 的治疗标准带来重大进展。
试验注册:ClinicalTrials.gov 标识符:NCT03443024。