Domondon Mark, Polina Iuliia, Nikiforova Anna B, Sultanova Regina F, Kruger Claudia, Vasileva Valeriia Y, Fomin Mikhail V, Beeson Gyda C, Nieminen Anna-Liisa, Smythe Nancy, Maldonado Eduardo N, Stadler Krisztian, Ilatovskaya Daria V
Department of Medicine, Division of Nephrology, Medical University of South Carolina, Charleston, SC, United States.
Institute of Theoretical and Experimental Biophysics, Pushchino, Russia.
Front Physiol. 2020 Feb 4;10:1588. doi: 10.3389/fphys.2019.01588. eCollection 2019.
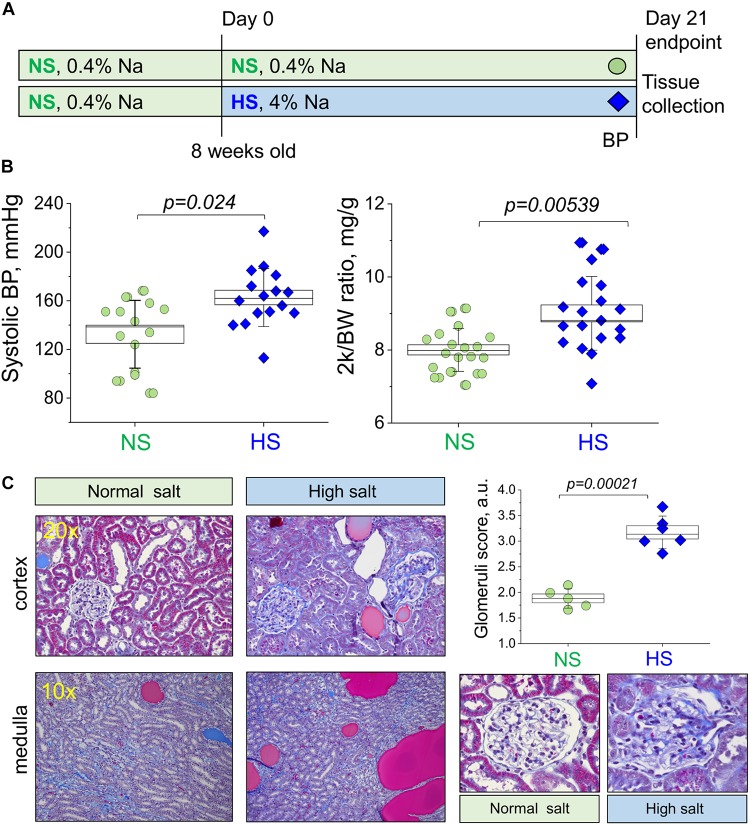
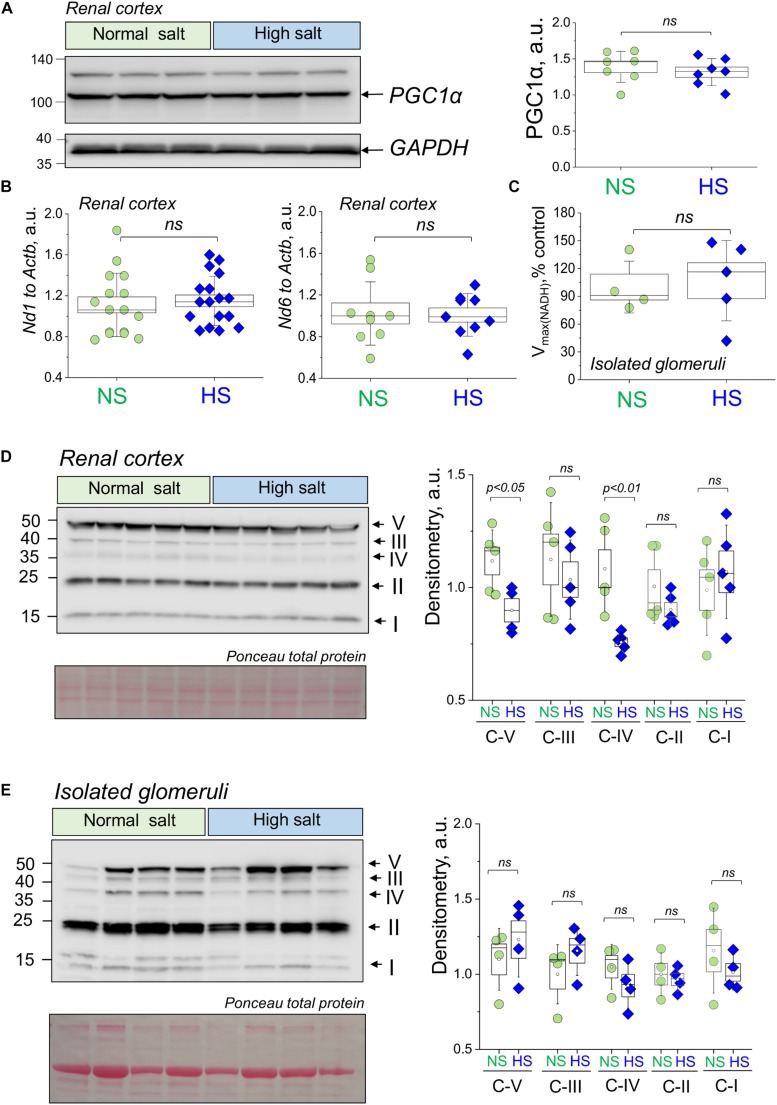
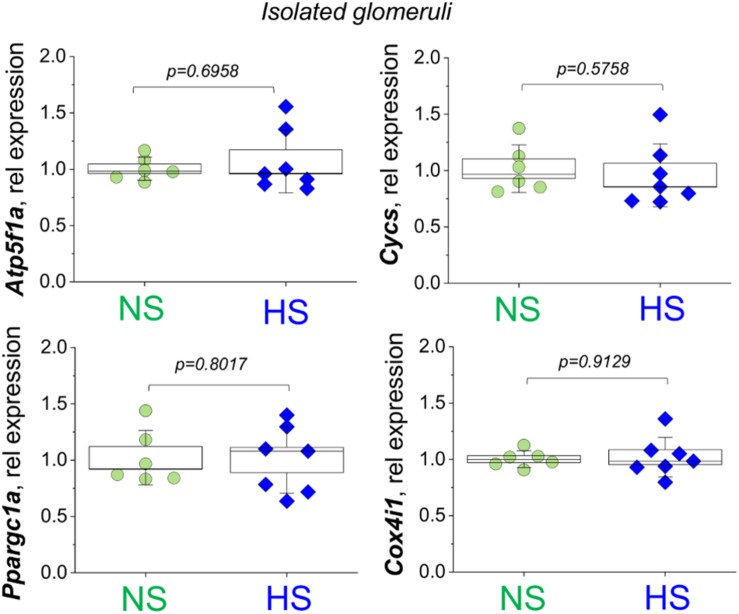
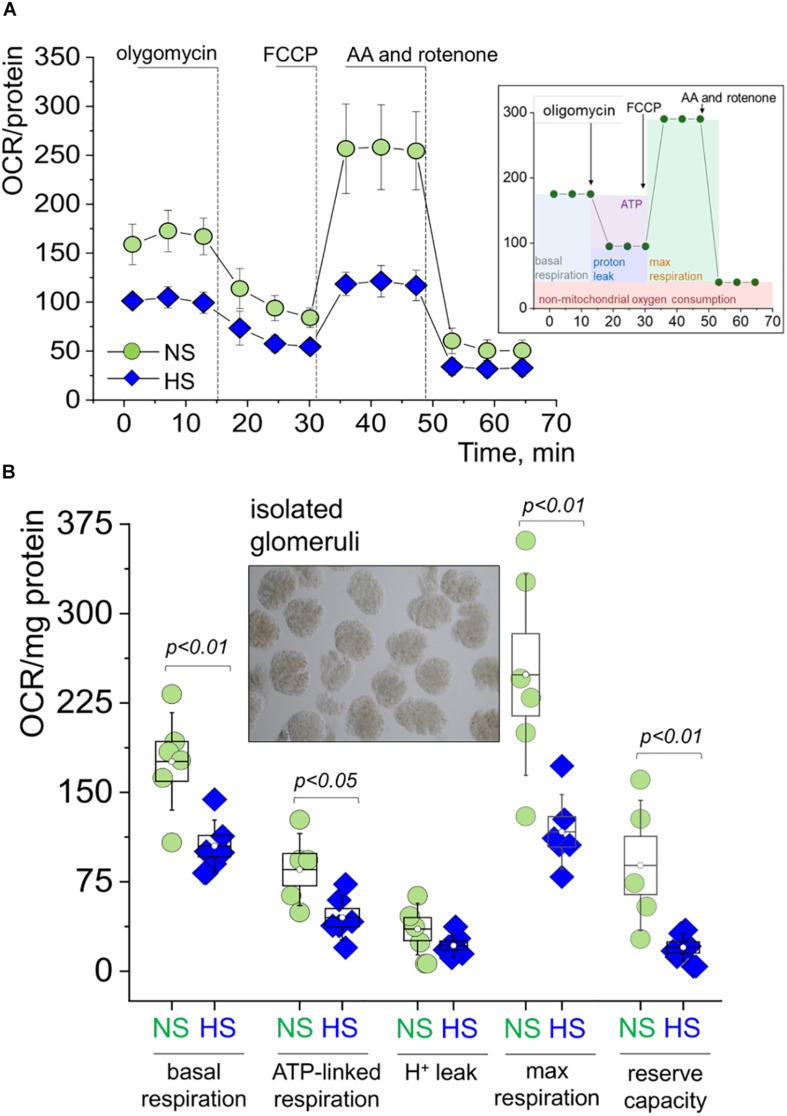
Salt-sensitive (SS) hypertension is accompanied with an early onset of proteinuria, which results from the loss of glomerular podocytes. Here, we hypothesized that glomerular damage in the SS hypertension occurs in part due to mitochondria dysfunction, and we used a unique model of freshly isolated glomeruli to test this hypothesis. In order to mimic SS hypertension, we used Dahl SS rats, an established animal model. Animals were fed a 0.4% NaCl (normal salt, NS) diet or challenged with a high salt (HS) 4% NaCl diet for 21 days to induce an increase in blood pressure (BP). Similar to previous studies, we found that HS diet caused renal hypertrophy, increased BP, glomerulosclerosis, and renal lesions such as fibrosis and protein casts. We did not observe changes in mitochondrial biogenesis in the renal cortex or isolated glomeruli fractions. However, Seahorse assay performed on freshly isolated glomeruli revealed that basal mitochondrial respiration, maximal respiration, and spare respiratory capacity were lower in the HS compared to the NS group. Using confocal imaging and staining for mitochondrial HO using mitoPY1, we detected an intensified response to an acute HO application in the podocytes of the glomeruli isolated from the HS diet fed group. TEM analysis showed that glomerular mitochondria from the HS diet fed group have structural abnormalities (swelling, enlargement, less defined cristae). Therefore, we report that glomerular mitochondria in SS hypertension are functionally and structurally defective, and this impairment could eventually lead to loss of podocytes and proteinuria. Thus, the glomerular-mitochondria axis can be targeted in novel treatment strategies for hypertensive glomerulosclerosis.
盐敏感性(SS)高血压伴有蛋白尿的早期发作,这是由肾小球足细胞丢失所致。在此,我们假设SS高血压中的肾小球损伤部分是由于线粒体功能障碍引起的,并且我们使用了新鲜分离肾小球的独特模型来验证这一假设。为了模拟SS高血压,我们使用了已建立的动物模型—— Dahl SS大鼠。动物被喂食0.4% NaCl(正常盐,NS)饮食或用4% NaCl高盐(HS)饮食刺激21天以诱导血压(BP)升高。与先前的研究相似,我们发现HS饮食导致肾脏肥大、血压升高、肾小球硬化以及肾损伤,如纤维化和蛋白管型。我们未观察到肾皮质或分离的肾小球部分中线粒体生物发生的变化。然而,对新鲜分离的肾小球进行的海马实验显示,与NS组相比,HS组的基础线粒体呼吸、最大呼吸和备用呼吸能力较低。使用共聚焦成像并用mitoPY1对线粒体血红素加氧酶(HO)进行染色,我们检测到在喂食HS饮食组分离的肾小球足细胞中,对急性HO应用的反应增强。透射电子显微镜(TEM)分析表明,喂食HS饮食组的肾小球线粒体存在结构异常(肿胀、增大、嵴不清晰)。因此,我们报告SS高血压中的肾小球线粒体在功能和结构上存在缺陷,这种损伤最终可能导致足细胞丢失和蛋白尿。因此,肾小球 - 线粒体轴可作为高血压性肾小球硬化新治疗策略的靶点。