Summers Kim M, Bush Stephen J, Wu Chunlei, Su Andrew I, Muriuki Charity, Clark Emily L, Finlayson Heather A, Eory Lel, Waddell Lindsey A, Talbot Richard, Archibald Alan L, Hume David A
Mater Research Institute-University of Queensland, Translational Research Institute, Woolloongabba, QLD, Australia.
Nuffield Department of Clinical Medicine, University of Oxford, Oxford, United Kingdom.
Front Genet. 2020 Feb 14;10:1355. doi: 10.3389/fgene.2019.01355. eCollection 2019.
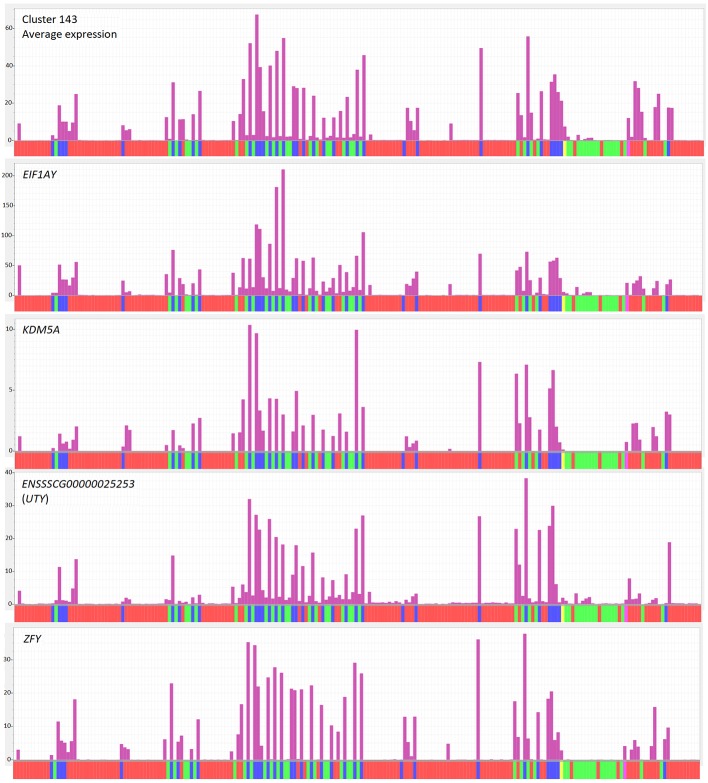
The domestic pig () is both an economically important livestock species and a model for biomedical research. Two highly contiguous pig reference genomes have recently been released. To support functional annotation of the pig genomes and comparative analysis with large human transcriptomic data sets, we aimed to create a pig gene expression atlas. To achieve this objective, we extended a previous approach developed for the chicken. We downloaded RNAseq data sets from public repositories, down-sampled to a common depth, and quantified expression against a reference transcriptome using the mRNA quantitation tool, Kallisto. We then used the network analysis tool Graphia to identify clusters of transcripts that were coexpressed across the merged data set. Consistent with the principle of guilt-by-association, we identified coexpression clusters that were highly tissue or cell-type restricted and contained transcription factors that have previously been implicated in lineage determination. Other clusters were enriched for transcripts associated with biological processes, such as the cell cycle and oxidative phosphorylation. The same approach was used to identify coexpression clusters within RNAseq data from multiple individual liver and brain samples, highlighting cell type, process, and region-specific gene expression. Evidence of conserved expression can add confidence to assignment of orthology between pig and human genes. Many transcripts currently identified as novel genes with ENSSSCG or LOC IDs were found to be coexpressed with annotated neighbouring transcripts in the same orientation, indicating they may be products of the same transcriptional unit. The meta-analytic approach to utilising public RNAseq data is extendable to include new data sets and new species and provides a framework to support the Functional Annotation of Animals Genomes (FAANG) initiative.
家猪()既是一种具有重要经济价值的家畜物种,也是生物医学研究的模型。最近发布了两个高度连续的猪参考基因组。为了支持猪基因组的功能注释以及与大量人类转录组数据集的比较分析,我们旨在创建一个猪基因表达图谱。为实现这一目标,我们扩展了先前为鸡开发的方法。我们从公共数据库下载RNAseq数据集,下采样到共同深度,并使用mRNA定量工具Kallisto针对参考转录组定量表达。然后,我们使用网络分析工具Graphia识别在合并数据集中共表达的转录本簇。与关联有罪原则一致,我们识别出高度受组织或细胞类型限制的共表达簇,其中包含先前与谱系确定有关的转录因子。其他簇富含与生物过程相关的转录本,如细胞周期和氧化磷酸化。我们使用相同的方法在来自多个个体肝脏和大脑样本的RNAseq数据中识别共表达簇,突出了细胞类型、过程和区域特异性基因表达。保守表达的证据可以增加猪和人类基因之间直系同源性分配的可信度。目前许多被鉴定为具有ENSSSCG或LOC ID的新基因的转录本,被发现与注释的相邻转录本以相同方向共表达,表明它们可能是同一转录单元的产物。利用公共RNAseq数据的元分析方法可扩展到包括新数据集和新物种,并为支持动物基因组功能注释(FAANG)计划提供了一个框架。