Dardis Andrea, Zampieri Stefania, Gellera Cinzia, Carrozzo Rosalba, Cattarossi Silvia, Peruzzo Paolo, Dariol Rosalia, Sechi Annalisa, Deodato Federica, Caccia Claudio, Verrigni Daniela, Gasperini Serena, Fiumara Agata, Fecarotta Simona, Carecchio Miryam, Filosto Massimiliano, Santoro Lucia, Borroni Barbara, Bordugo Andrea, Brancati Francesco, Russo Cinzia V, Di Rocco Maja, Toscano Antonio, Scarpa Maurizio, Bembi Bruno
Regional Coordinator Centre for Rare Diseases, University Hospital of Udine, 33100 Udine, Italy.
Unit of Genetics of Neurodegenerative and Metabolic Diseases Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milan, Italy.
J Clin Med. 2020 Mar 3;9(3):679. doi: 10.3390/jcm9030679.
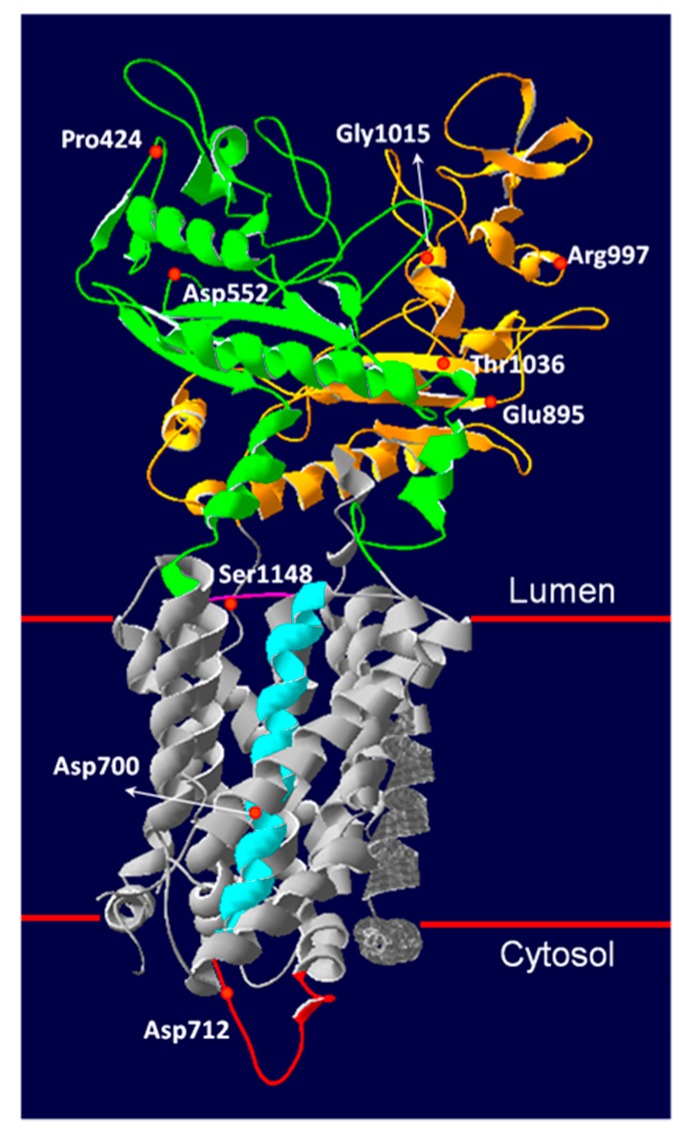
Niemann-Pick type C (NPC) disease is an autosomal recessive lysosomal storage disorder caused by mutations in or genes. In 2009, the molecular characterization of 44 NPC Italian patients has been published. Here, we present an update of the genetic findings in 105 Italian NPC patients belonging to 83 unrelated families (77 NPC1 and 6 NPC2). and genes were studied following an algorithm recently published. Eighty-four different and five alleles were identified. Only two alleles remained non detected. Sixty-two percent of alleles were due to missense variants. The most frequent mutation was the p.F284Lfs*26 (5.8% of the alleles). All mutations were found in the homozygous state, and all but one was severe. Among newly diagnosed patients, 18 novel mutations were identified. The pathogenic nature of 7/9 missense alleles and 3/4 intronic variants was confirmed by filipin staining and NPC1 protein analysis or mRNA expression in patient's fibroblasts. Taken together, our previous published data and new results provide an overall picture of the molecular characteristics of NPC patients diagnosed so far in Italy.
尼曼-皮克C型(NPC)病是一种常染色体隐性溶酶体贮积症,由 或 基因的突变引起。2009年,已发表了44例意大利NPC患者的分子特征。在此,我们报告了105例意大利NPC患者(来自83个无亲缘关系的家庭,其中77例为NPC1型,6例为NPC2型)的基因研究新结果。按照最近发表的一种算法对 和 基因进行了研究。共鉴定出84种不同的 等位基因和5种 等位基因。仅2种 等位基因未被检测到。62%的 等位基因是错义变异。最常见的 突变是p.F284Lfs*26(占等位基因的5.8%)。所有 突变均为纯合状态,除1例以外均为严重突变。在新诊断的患者中,鉴定出18种新的 突变。通过对患者成纤维细胞进行荧光素染色、NPC1蛋白分析或mRNA表达,证实了7/9个错义等位基因和3/4个内含子变异的致病性质。综合来看,我们之前发表的数据和新结果提供了意大利目前已诊断NPC患者分子特征的全貌。