Group of Inverse Problems, Optimization and Machine Learning, Department of Mathematics, University of Oviedo, C/ Federico García-Lorca, 18, 33007, Oviedo, Spain.
Department of Informatics and Computer Science, University of Oviedo, C/ Federico García-Lorca, 18, 33007, Oviedo, Spain.
BMC Bioinformatics. 2020 Mar 11;21(Suppl 2):89. doi: 10.1186/s12859-020-3356-6.
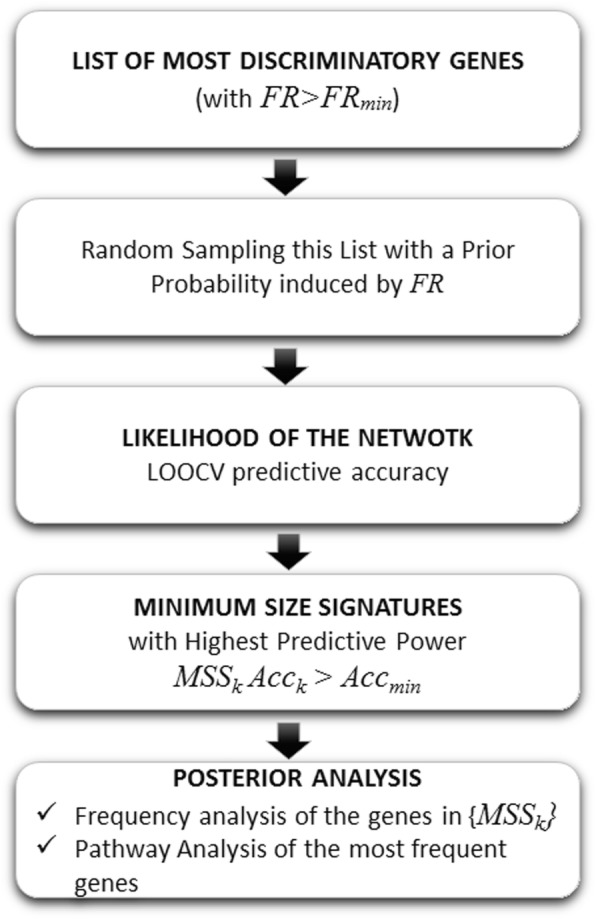
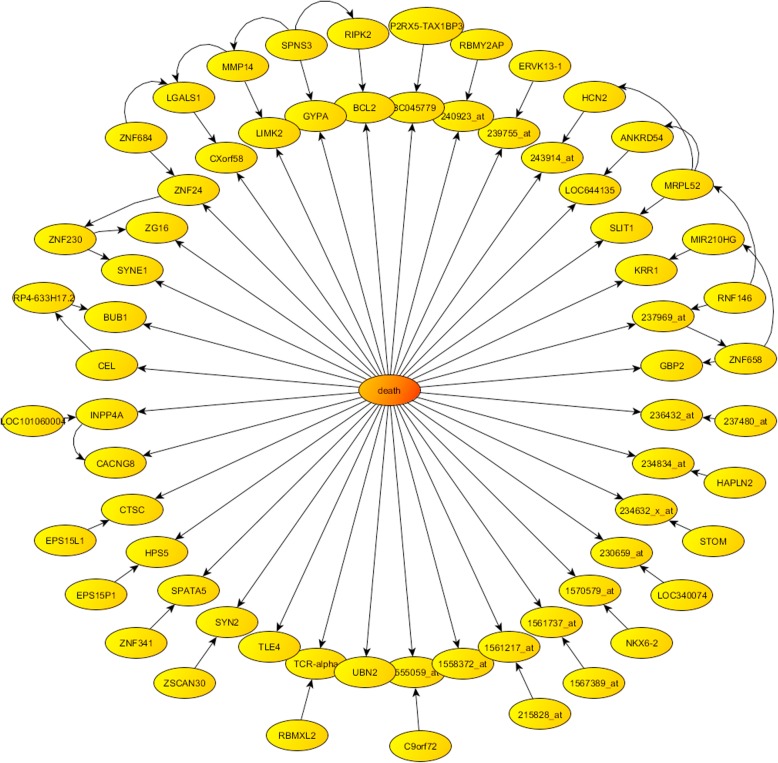
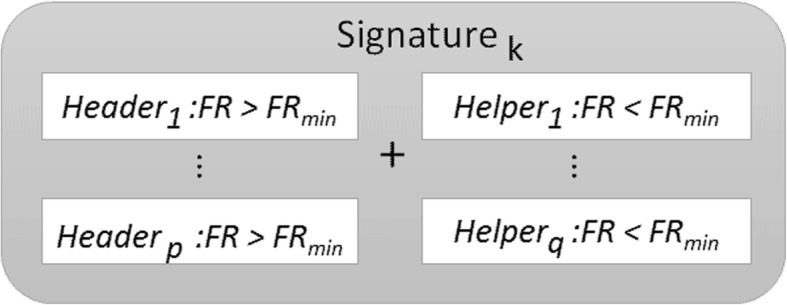
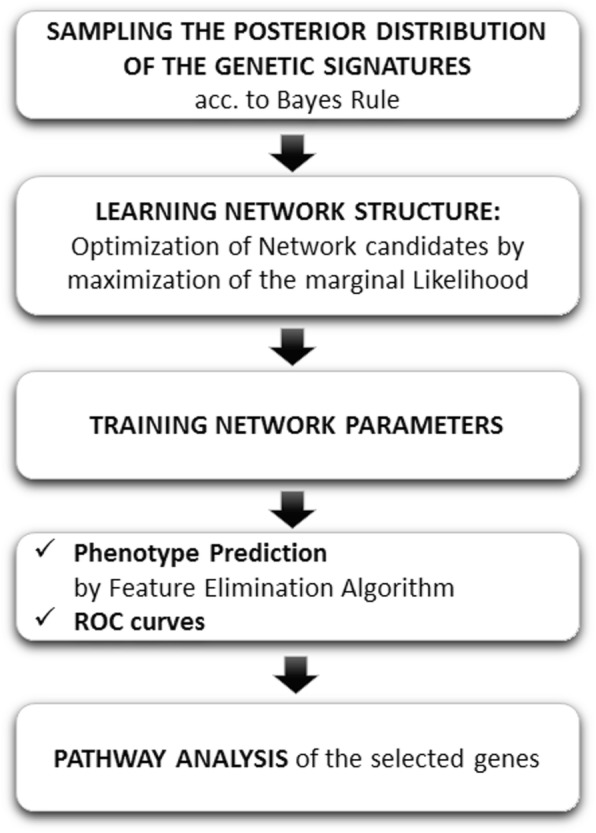
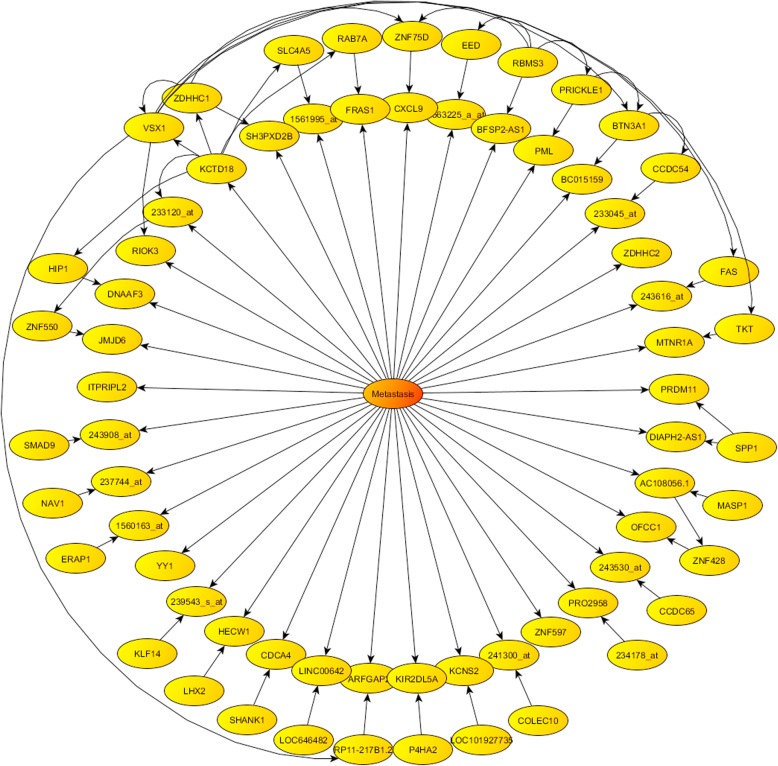
Phenotype prediction problems are usually considered ill-posed, as the amount of samples is very limited with respect to the scrutinized genetic probes. This fact complicates the sampling of the defective genetic pathways due to the high number of possible discriminatory genetic networks involved. In this research, we outline three novel sampling algorithms utilized to identify, classify and characterize the defective pathways in phenotype prediction problems, such as the Fisher's ratio sampler, the Holdout sampler and the Random sampler, and apply each one to the analysis of genetic pathways involved in tumor behavior and outcomes of triple negative breast cancers (TNBC). Altered biological pathways are identified using the most frequently sampled genes and are compared to those obtained via Bayesian Networks (BNs).
Random, Fisher's ratio and Holdout samplers were more accurate and robust than BNs, while providing comparable insights about disease genomics.
The three samplers tested are good alternatives to Bayesian Networks since they are less computationally demanding algorithms. Importantly, this analysis confirms the concept of "biological invariance" since the altered pathways should be independent of the sampling methodology and the classifier used for their inference. Nevertheless, still some modifications are needed in the Bayesian networks to be able to sample correctly the uncertainty space in phenotype prediction problems, since the probabilistic parameterization of the uncertainty space is not unique and the use of the optimum network might falsify the pathways analysis.
表型预测问题通常被认为是不适定的,因为与被研究的遗传探针相比,样本量非常有限。由于涉及的可能具有鉴别力的遗传网络数量众多,这一事实使得有缺陷的遗传途径的抽样变得复杂。在这项研究中,我们概述了三种新的采样算法,用于识别、分类和表征表型预测问题中的有缺陷的途径,如 Fisher 比率采样器、Holdout 采样器和随机采样器,并将每种算法应用于分析肿瘤行为和三阴性乳腺癌 (TNBC) 结果中涉及的遗传途径。使用最常采样的基因来识别改变的生物途径,并将其与通过贝叶斯网络 (BN) 获得的途径进行比较。
随机、Fisher 比率和 Holdout 采样器比 BNs 更准确和稳健,同时提供了关于疾病基因组学的可比见解。
测试的三种采样器是贝叶斯网络的良好替代品,因为它们的计算要求较低。重要的是,这项分析证实了“生物学不变性”的概念,因为改变的途径应该独立于采样方法和用于推断它们的分类器。然而,在贝叶斯网络中仍然需要进行一些修改,以便能够正确地对表型预测问题中的不确定性空间进行采样,因为不确定性空间的概率参数化不是唯一的,并且使用最优网络可能会使途径分析产生偏差。