Department of Biotechnology and Bioindustry Sciences, National Cheng Kung University, Tainan, 701, Taiwan.
Molecular Diagnostic Laboratory, Department of Pathology, National Cheng Kung University Hospital, Tainan, Taiwan.
BMC Bioinformatics. 2020 Mar 14;21(1):105. doi: 10.1186/s12859-020-3445-6.
Illumina sequencing of a marker gene is popular in metagenomic studies. However, Illumina paired-end (PE) reads sometimes cannot be merged into single reads for subsequent analysis. When mergeable PE reads are limited, one can simply use only first reads for taxonomy annotation, but that wastes information in the second reads. Presumably, including second reads should improve taxonomy annotation. However, a rigorous investigation of how best to do this and how much can be gained has not been reported.
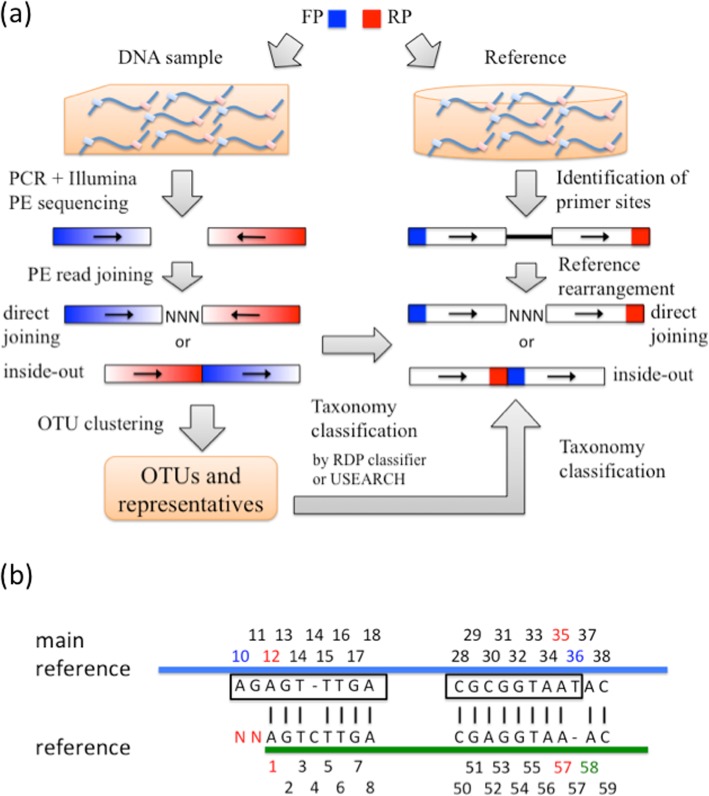
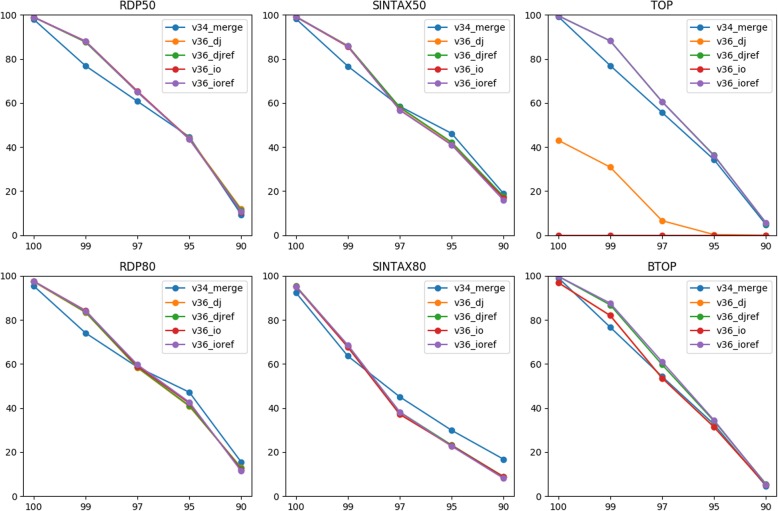
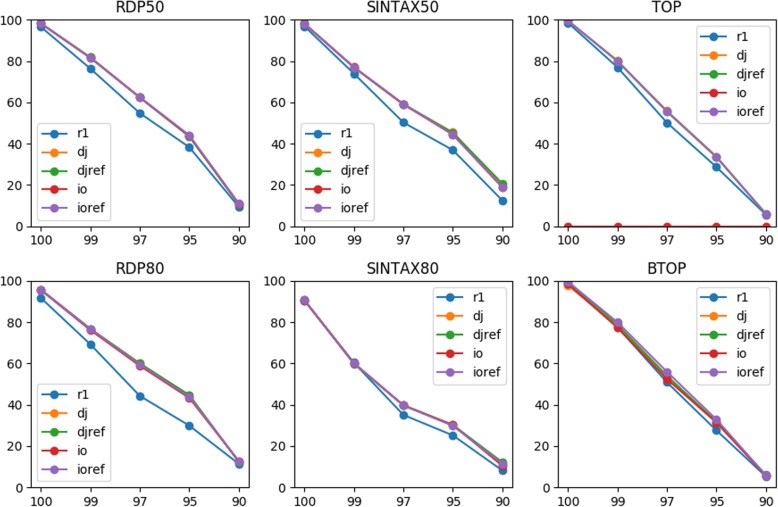
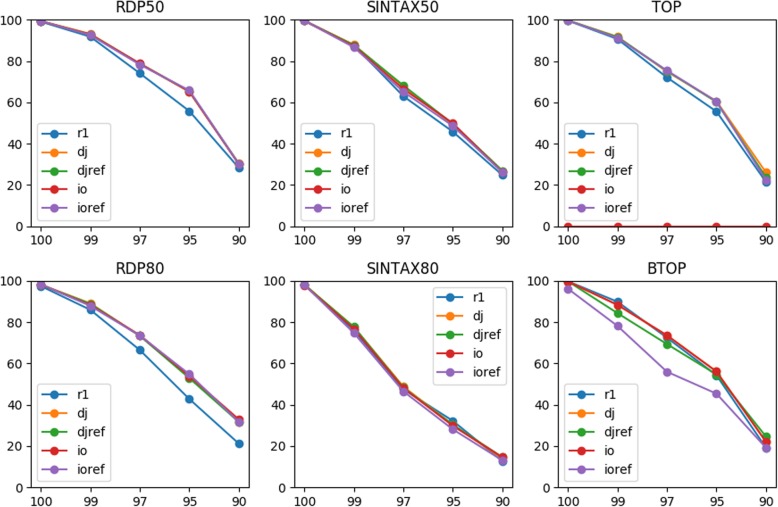
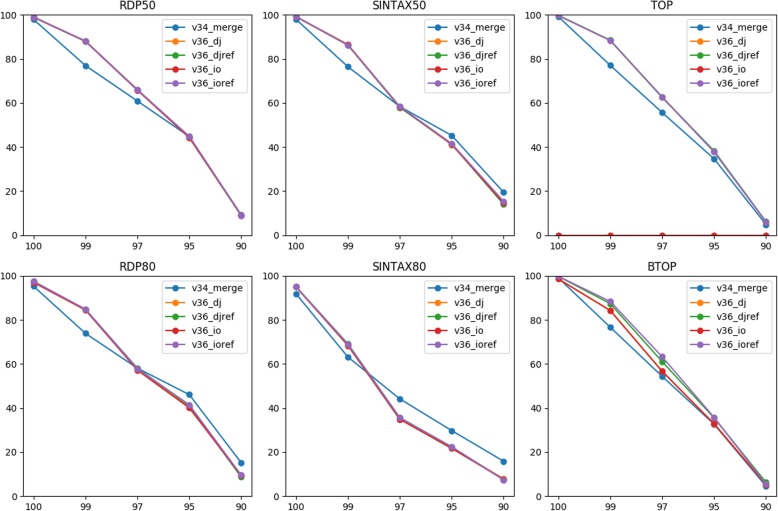
We evaluated two methods of joining as opposed to merging PE reads into single reads for taxonomy annotation using simulated data with sequencing errors. Our rigorous evaluation involved several top classifiers (RDP classifier, SINTAX, and two alignment-based methods) and realistic benchmark datasets. For most classifiers, read joining ameliorated the impact of sequencing errors and improved the accuracy of taxonomy predictions. For alignment-based top-hit classifiers, rearranging the reference sequences is recommended to avoid improper alignments of joined reads. For word-counting classifiers, joined reads could be compared to the original reference for classification. We also applied read joining to our own real MiSeq PE data of nasal microbiota of asthmatic children. Before joining, trimming low quality bases was necessary for optimizing taxonomy annotation and sequence clustering. We then showed that read joining increased the amount of effective data for taxonomy annotation. Using these joined trimmed reads, we were able to identify two promising bacterial genera that might be associated with asthma exacerbation.
When mergeable PE reads are limited, joining them into single reads for taxonomy annotation is always recommended. Reference sequences may need to be rearranged accordingly depending on the classifier. Read joining also relaxes the constraint on primer selection, and thus may unleash the full capacity of Illumina PE data for taxonomy annotation. Our work provides guidance for fully utilizing PE data of a marker gene when mergeable reads are limited.
Illumina 测序在宏基因组研究中很受欢迎。然而,Illumina 配对末端(PE)reads 有时无法合并为单读用于后续分析。当可合并的 PE reads 有限时,人们可以简单地仅使用第一读进行分类注释,但这会浪费第二读中的信息。推测,包括第二读应该可以改善分类注释。然而,如何最好地做到这一点以及可以获得多少收益,尚未有报道。
我们使用具有测序错误的模拟数据评估了两种将 PE reads 合并为单读进行分类注释的方法。我们的严格评估涉及几种顶级分类器(RDP 分类器、SINTAX 和两种基于比对的方法)和现实的基准数据集。对于大多数分类器,读取合并改善了测序错误的影响并提高了分类预测的准确性。对于基于比对的顶级命中分类器,建议重新排列参考序列以避免合并读取的不当比对。对于基于单词计数的分类器,可以将合并的读取与原始参考进行比较以进行分类。我们还将读取合并应用于我们自己的真实 MiSeq PE 数据的哮喘儿童鼻腔微生物组。在合并之前,需要修剪低质量的碱基来优化分类注释和序列聚类。然后,我们表明,读取合并增加了分类注释的有效数据量。使用这些合并的修剪读取,我们能够鉴定出两个可能与哮喘加重相关的有前途的细菌属。
当可合并的 PE reads 有限时,建议将它们合并为单读进行分类注释。根据分类器的不同,可能需要相应地重新排列参考序列。读取合并还放宽了对引物选择的限制,从而可能释放 Illumina PE 数据用于分类注释的全部容量。我们的工作为充分利用有限的可合并读时标记基因的 PE 数据提供了指导。