Institute of Oceanology, Chinese Academy of Sciences, Qingdao, 266071, China.
Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, 266071, China.
BMC Genomics. 2020 Mar 17;21(1):240. doi: 10.1186/s12864-020-6642-9.
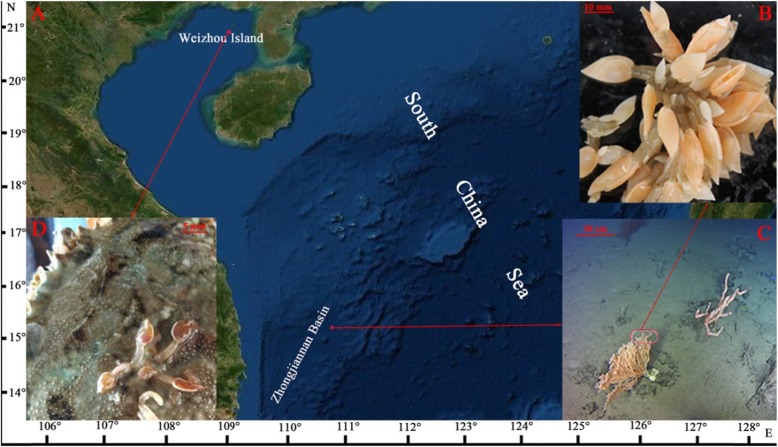
Barnacles are specialized marine organisms that differ from other crustaceans in possession of a calcareous shell, which is attached to submerged surfaces. Barnacles have a wide distribution, mostly in the intertidal zone and shallow waters, but a few species inhabit the deep-sea floor. It is of interest to investigate how such sessile crustaceans became adapted to extreme deep-sea environments. We sequenced the transcriptomes of a deep-sea barnacle, Glyptelasma gigas collected at a depth of 731 m from the northern area of the Zhongjiannan Basin, and a shallow-water coordinal relative, Octolasmis warwicki. The purpose of this study was to provide genetic resources for investigating adaptation mechanisms of deep-sea barnacles.
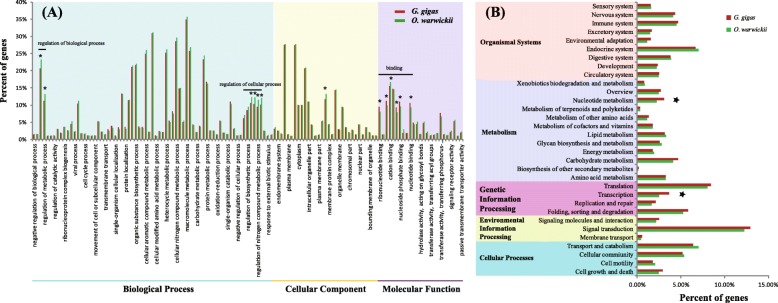
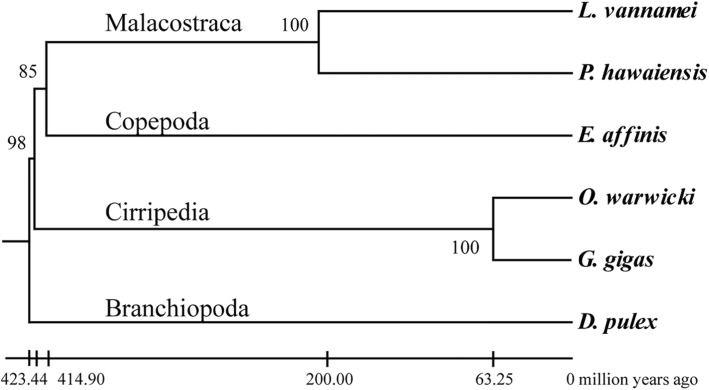
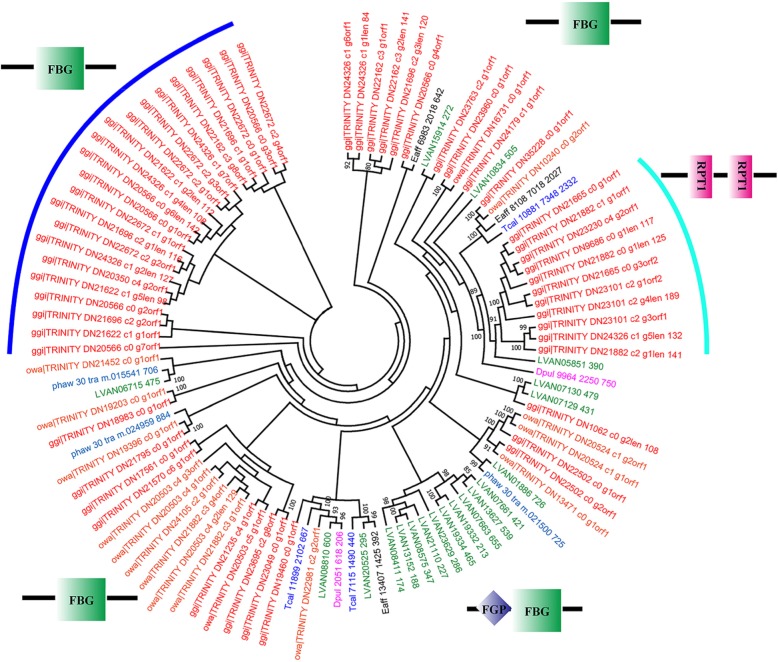
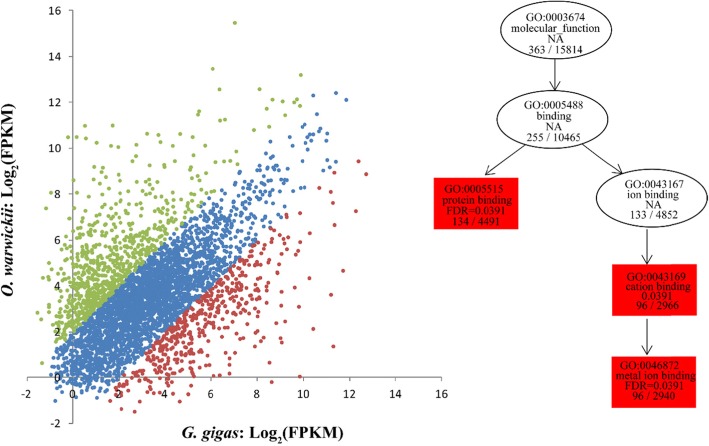
Totals of 62,470 and 51,585 unigenes were assembled for G. gigas and O. warwicki, respectively, and functional annotation of these unigenes was made using public databases. Comparison of the protein-coding genes between the deep- and shallow-water barnacles, and with those of four other shallow-water crustaceans, revealed 26 gene families that had experienced significant expansion in G. gigas. Functional annotation showed that these expanded genes were predominately related to DNA repair, signal transduction and carbohydrate metabolism. Base substitution analysis on the 11,611 single-copy orthologs between G. gigas and O. warwicki indicated that 25 of them were distinctly positive selected in the deep-sea barnacle, including genes related to transcription, DNA repair, ligand binding, ion channels and energy metabolism, potentially indicating their importance for survival of G. gigas in the deep-sea environment.
The barnacle G. gigas has adopted strategies of expansion of specific gene families and of positive selection of key genes to counteract the negative effects of high hydrostatic pressure, hypoxia, low temperature and food limitation on the deep-sea floor. These expanded gene families and genes under positive selection would tend to enhance the capacities of G. gigas for signal transduction, genetic information processing and energy metabolism, and facilitate networks for perceiving and responding physiologically to the environmental conditions in deep-sea habitats. In short, our results provide genomic evidence relating to deep-sea adaptation of G. gigas, which provide a basis for further biological studies of sessile crustaceans in the deep sea.
藤壶是一种特殊的海洋生物,与其他甲壳类动物不同,它们拥有钙质外壳,附着在水下物体上。藤壶分布广泛,主要生活在潮间带和浅水区,但也有少数物种栖息在深海海底。研究这些固着甲壳类动物如何适应极端深海环境非常有趣。我们对从中印尼海盆北部水深 731 米处采集的深海藤壶 Glyptelasma gigas 和浅水区的亲缘种 Octolasmis warwicki 进行了转录组测序。本研究的目的是为研究深海藤壶的适应机制提供遗传资源。
分别为 G. gigas 和 O. warwicki 组装了 62470 和 51585 条 unigenes,并使用公共数据库对这些 unigenes 进行了功能注释。将深海和浅海藤壶的蛋白质编码基因与另外四种浅海甲壳动物的基因进行比较,发现 G. gigas 中有 26 个基因家族发生了显著扩张。功能注释表明,这些扩张的基因主要与 DNA 修复、信号转导和碳水化合物代谢有关。对 G. gigas 和 O. warwicki 之间的 11611 个单拷贝直系同源基因进行碱基替换分析表明,其中 25 个在深海藤壶中明显受到正选择,包括与转录、DNA 修复、配体结合、离子通道和能量代谢相关的基因,这可能表明它们对 G. gigas 在深海环境中的生存至关重要。
藤壶 G. gigas 采用了特定基因家族扩张和关键基因正选择的策略,以抵消深海海底高压、缺氧、低温和食物限制对其的负面影响。这些扩张的基因家族和受到正选择的基因可能会增强 G. gigas 进行信号转导、遗传信息处理和能量代谢的能力,并促进其感知和生理响应深海栖息地环境条件的网络。总之,我们的研究结果为 G. gigas 的深海适应提供了基因组证据,为进一步研究深海固着甲壳类动物提供了基础。