Jiang Li, Zhang Mengmeng, Wang Sixue, Han Yuanyuan, Fang Xiaoling
Department of Obstetrics and Gynecology, The Second Xiangya Hospital, Central South University, Changsha, Hunan, China.
Center of Tree Shrew Germplasm Resources, Institute of Medical Biology, Chinese Academy of Medical Sciences and Peking Union Medical College, Kunming, Yunnan, China.
PeerJ. 2020 Mar 5;8:e8730. doi: 10.7717/peerj.8730. eCollection 2020.
To identify the common and specific molecular mechanisms of three well-defined subtypes of endometriosis (EMs): ovarian endometriosis (OE), peritoneal endometriosis (PE), and deep infiltrating endometriosis (DIE).
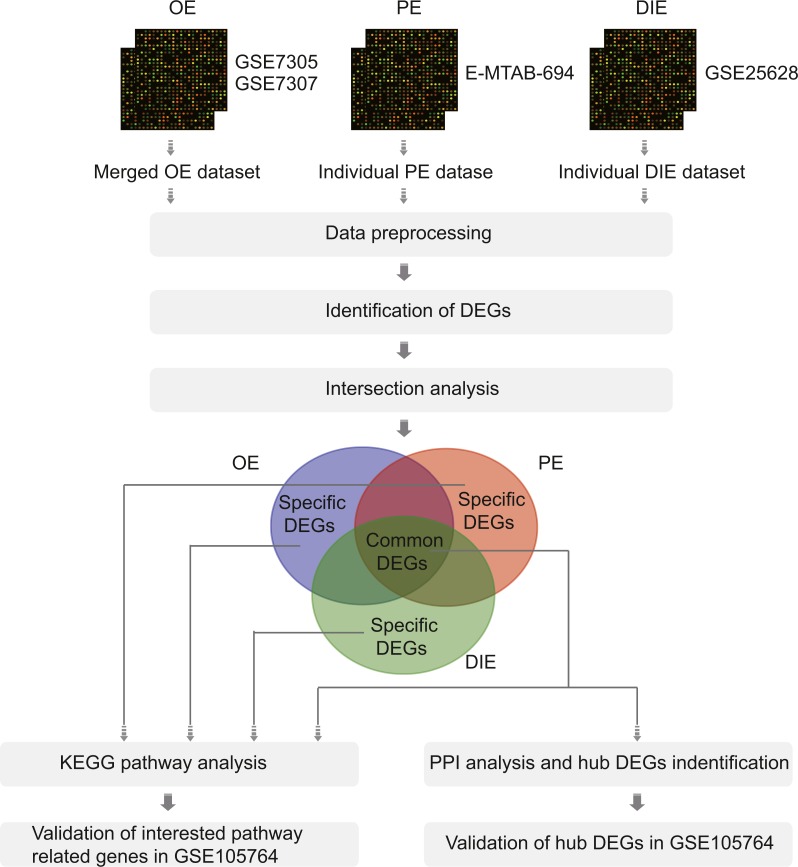
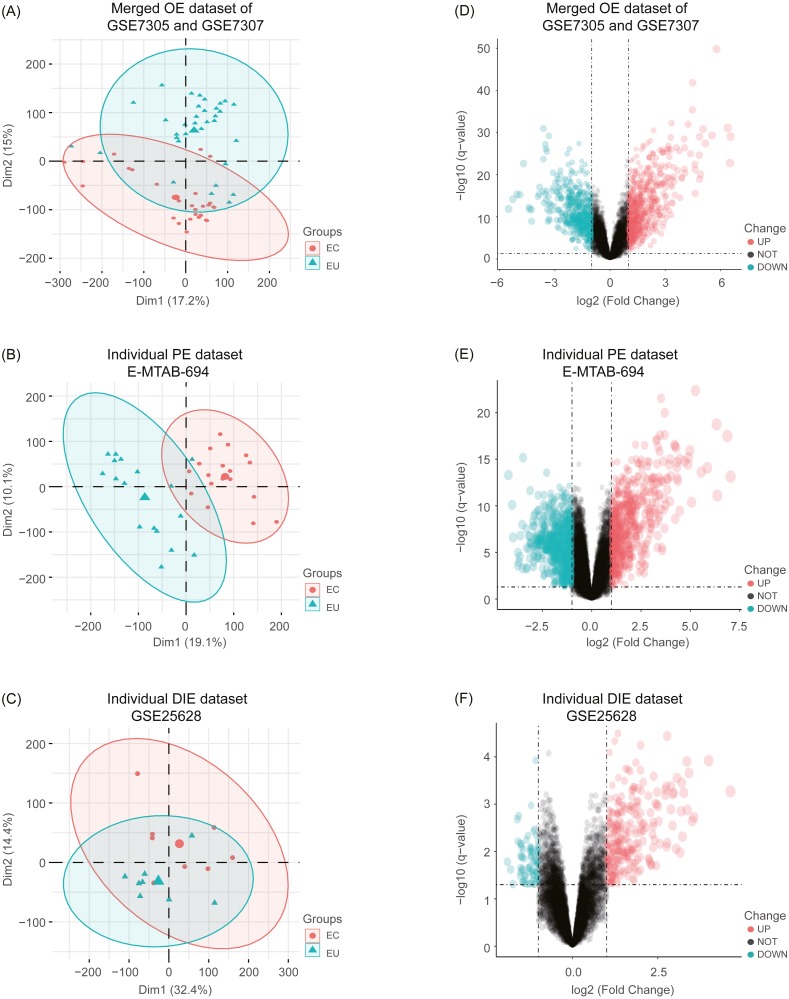
Four microarray datasets: GSE7305 and GSE7307 for OE, E-MTAB-694 for PE, and GSE25628 for DIE were downloaded from public databases and conducted to compare ectopic lesions (EC) with eutopic endometrium (EU) from EMs patients. Differentially expressed genes (DEGs) identified by limma package were divided into two parts: common DEGs among three subtypes and specific DEGs in each subtype, both of which were subsequently performed with the Kyoto Encyclopedia of Genes (KEGG) pathway enrichment analysis. The protein-protein interaction (PPI) network was constructed by common DEGs and five hub genes were screened out from the PPI network. Besides, these five hub genes together with selected interested pathway-related genes were further validated in an independent OE RNA-sequencing dataset GSE105764.
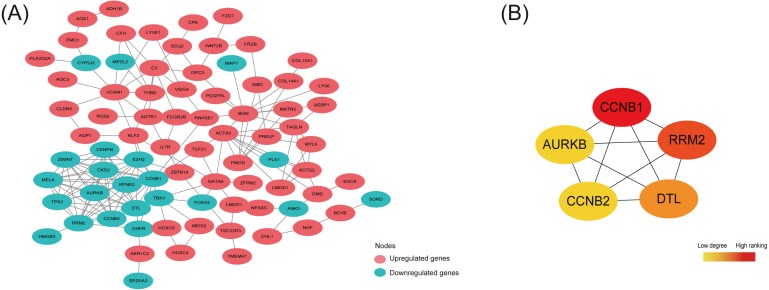
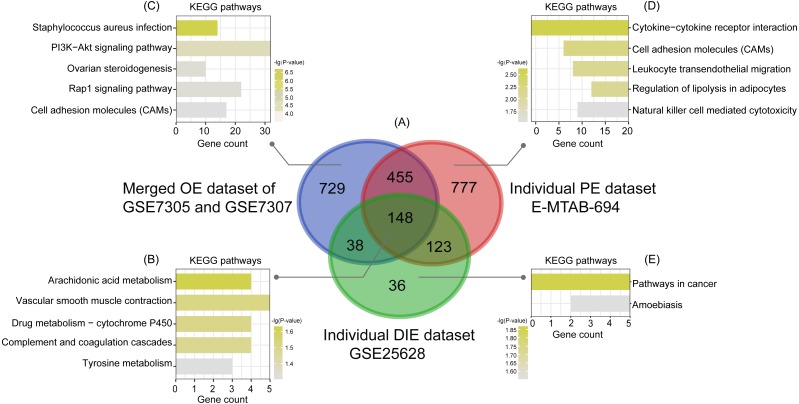
A total of 54 EC samples from three EMs subtypes (OE, PE, DIE) and 58 EU samples were analyzed, from which we obtained 148 common DEGs among three subtypes, and 729 specific DEGs in OE, 777 specific DEGs in PE and 36 specific DEGs in DIE. The most enriched pathway of 148 shared DEGs was arachidonic acid (AA) metabolism, in which most genes were up-regulated in EC, indicating inflammation was the most common pathogenesis of three subtypes. Besides, five hub genes AURKB, RRM2, DTL, CCNB1, CCNB2 identified from the PPI network constructed by 148 shared DEGs were all associated with cell cycle and mitosis, and down-regulated in EC, suggesting a slow and controlled proliferation in ectopic lesions. The KEGG pathway analysis of specific DEGs in each subtype revealed that abnormal ovarian steroidogenesis was a prominent feature in OE; OE and DIE seems to be at more risk of malignant development since both of their specific DEGs were enriched in the pathways in cancer, though enriched genes were different, while PE tended to be more associated with dysregulated peritoneal immune and inflammatory microenvironment.
By integrated bioinformatic analysis, we explored common and specific molecular signatures among different subtypes of endometriosis: activated arachidonic acid (AA) metabolism-related inflammatory process and a slow and controlled proliferation in ectopic lesions were common features in OE, PE and DIE; OE and DIE seemed to be at more risk of malignant development while PE tended to be more associated with dysregulated peritoneal immune and inflammatory microenvironment, all of which could deepen our perception of endometriosis.
确定三种明确的子宫内膜异位症(EMs)亚型的共同和特定分子机制:卵巢子宫内膜异位症(OE)、腹膜子宫内膜异位症(PE)和深部浸润性子宫内膜异位症(DIE)。
从公共数据库下载四个微阵列数据集:用于OE的GSE7305和GSE7307、用于PE的E-MTAB-694以及用于DIE的GSE25628,并进行分析以比较EMs患者的异位病灶(EC)和在位内膜(EU)。使用limma软件包鉴定出的差异表达基因(DEGs)分为两部分:三种亚型之间的共同DEGs和各亚型中的特定DEGs,随后对这两部分基因均进行京都基因与基因组百科全书(KEGG)通路富集分析。通过共同DEGs构建蛋白质-蛋白质相互作用(PPI)网络,并从PPI网络中筛选出五个枢纽基因。此外,在一个独立的OE RNA测序数据集GSE105764中进一步验证了这五个枢纽基因以及选定的感兴趣的通路相关基因。
共分析了来自三种EMs亚型(OE、PE、DIE)的54个EC样本和58个EU样本,从中我们获得了三种亚型之间的148个共同DEGs,以及OE中的729个特定DEGs、PE中的777个特定DEGs和DIE中的36个特定DEGs。148个共享DEGs中最富集的通路是花生四烯酸(AA)代谢,其中大多数基因在EC中上调,表明炎症是三种亚型最常见的发病机制。此外,从由148个共享DEGs构建的PPI网络中鉴定出的五个枢纽基因AURKB、RRM2、DTL、CCNB1、CCNB2均与细胞周期和有丝分裂相关,且在EC中下调,提示异位病灶中增殖缓慢且受到控制。对各亚型中特定DEGs的KEGG通路分析显示,异常的卵巢类固醇生成是OE的一个突出特征;OE和DIE似乎有更高的恶性发展风险,因为它们的特定DEGs均在癌症相关通路中富集,尽管富集的基因不同,而PE则更倾向于与腹膜免疫和炎症微环境失调相关。
通过综合生物信息学分析,我们探索了不同亚型子宫内膜异位症之间的共同和特定分子特征:激活的花生四烯酸(AA)代谢相关炎症过程以及异位病灶中增殖缓慢且受到控制是OE、PE和DIE的共同特征;OE和DIE似乎有更高的恶性发展风险,而PE则更倾向于与腹膜免疫和炎症微环境失调相关,所有这些都可以加深我们对子宫内膜异位症的认识。