Department of Earth and Planetary Sciences, University of California, Berkeley, California 94720, USA.
Graduate Program in Biophysical Sciences, University of Chicago, Chicago, Illinois 60637, USA.
Genome Res. 2020 Mar;30(3):315-333. doi: 10.1101/gr.258640.119. Epub 2020 Mar 18.
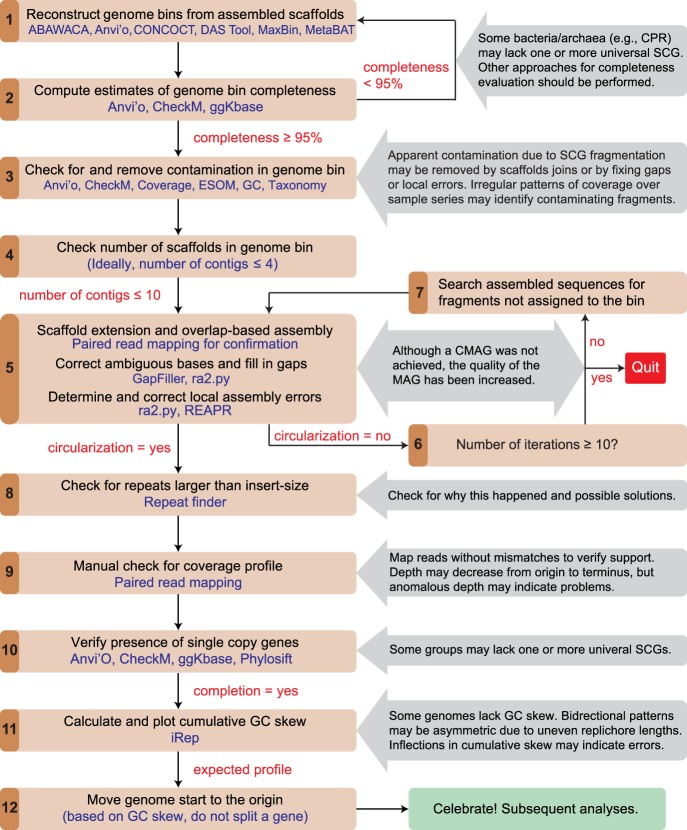
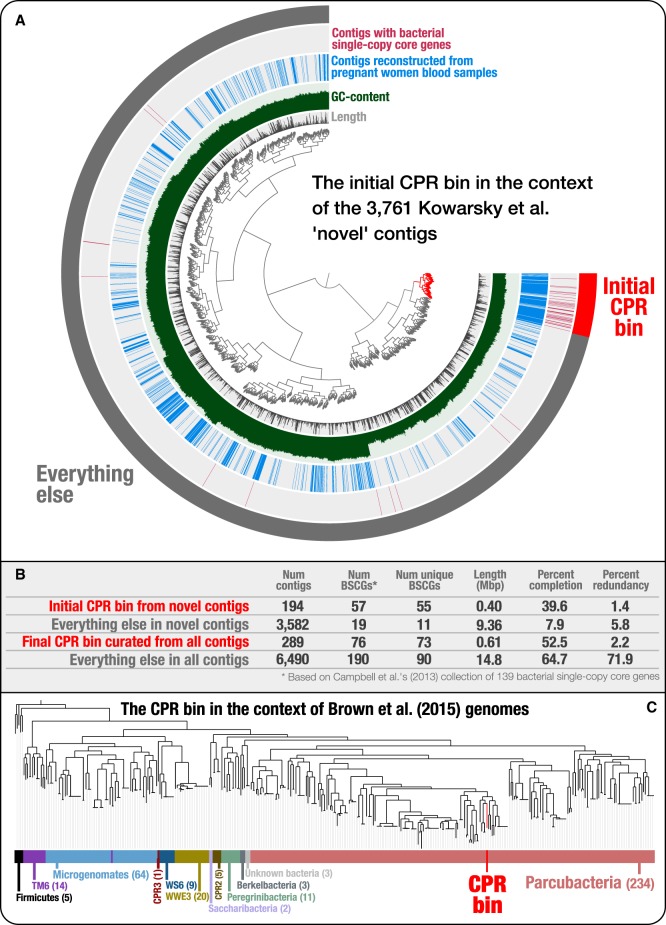
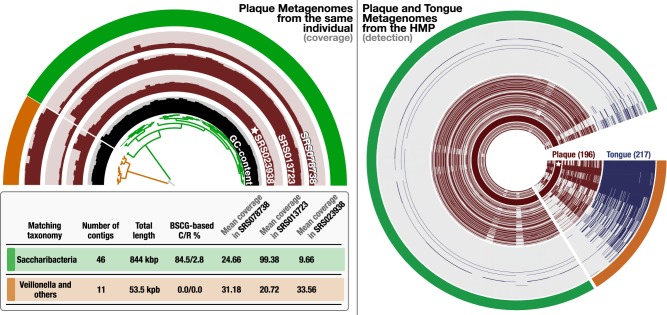
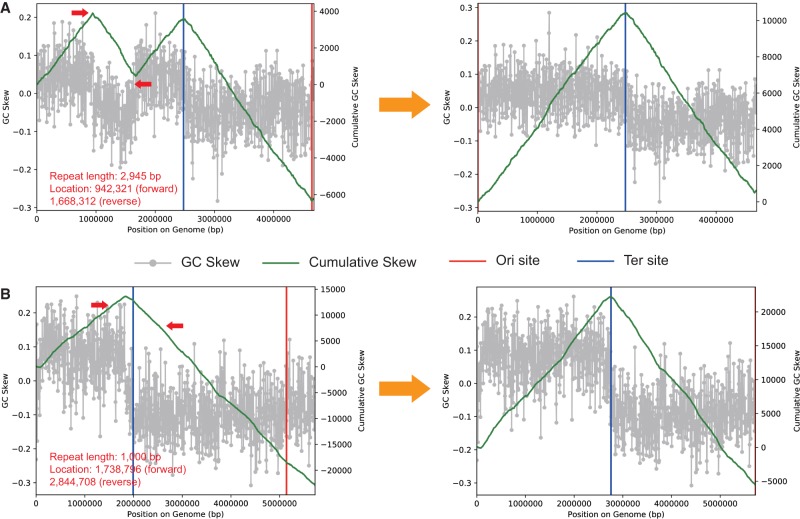
Genomes are an integral component of the biological information about an organism; thus, the more complete the genome, the more informative it is. Historically, bacterial and archaeal genomes were reconstructed from pure (monoclonal) cultures, and the first reported sequences were manually curated to completion. However, the bottleneck imposed by the requirement for isolates precluded genomic insights for the vast majority of microbial life. Shotgun sequencing of microbial communities, referred to initially as community genomics and subsequently as genome-resolved metagenomics, can circumvent this limitation by obtaining metagenome-assembled genomes (MAGs); but gaps, local assembly errors, chimeras, and contamination by fragments from other genomes limit the value of these genomes. Here, we discuss genome curation to improve and, in some cases, achieve complete (circularized, no gaps) MAGs (CMAGs). To date, few CMAGs have been generated, although notably some are from very complex systems such as soil and sediment. Through analysis of about 7000 published complete bacterial isolate genomes, we verify the value of cumulative GC skew in combination with other metrics to establish bacterial genome sequence accuracy. The analysis of cumulative GC skew identified potential misassemblies in some reference genomes of isolated bacteria and the repeat sequences that likely gave rise to them. We discuss methods that could be implemented in bioinformatic approaches for curation to ensure that metabolic and evolutionary analyses can be based on very high-quality genomes.
基因组是生物体内有关生物信息的一个重要组成部分;因此,基因组越完整,其信息量就越大。从历史上看,细菌和古菌的基因组是从纯(单克隆)培养物中重建的,最初报道的序列是经过手工精心整理完成的。然而,由于需要分离物的限制,这一过程成为了绝大多数微生物生命的基因组学研究的瓶颈。微生物群落的鸟枪法测序,最初被称为群落基因组学,随后又被称为基因组解析宏基因组学,通过获得宏基因组组装基因组(MAG)可以避免这一限制;但是,缺口、局部组装错误、嵌合体和来自其他基因组片段的污染限制了这些基因组的价值。在这里,我们讨论了基因组整理,以提高和在某些情况下实现完整的(环形化,无缺口)MAG(CMAG)。到目前为止,虽然已经生成了一些非常复杂的系统,如土壤和沉积物中的 CMAG,但数量仍然很少。通过对大约 7000 个已发表的完整细菌分离株基因组的分析,我们验证了累积 GC 倾斜度与其他指标相结合在确定细菌基因组序列准确性方面的价值。累积 GC 倾斜度的分析确定了一些分离细菌的参考基因组中可能存在的错误组装,并确定了可能导致这些错误组装的重复序列。我们讨论了可以在生物信息学方法中实施的方法,以确保代谢和进化分析可以基于非常高质量的基因组。