Gupta Ankur K, Stulajter Miko M, Shaidu Yusuf, Neaton Jeffrey B, de Jong Wibe A
Applied Mathematics and Computational Research Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States.
Department of Physics, University of California Berkeley, Berkeley, California 94720, United States.
ACS Omega. 2024 Sep 11;9(38):40269-40282. doi: 10.1021/acsomega.4c07434. eCollection 2024 Sep 24.
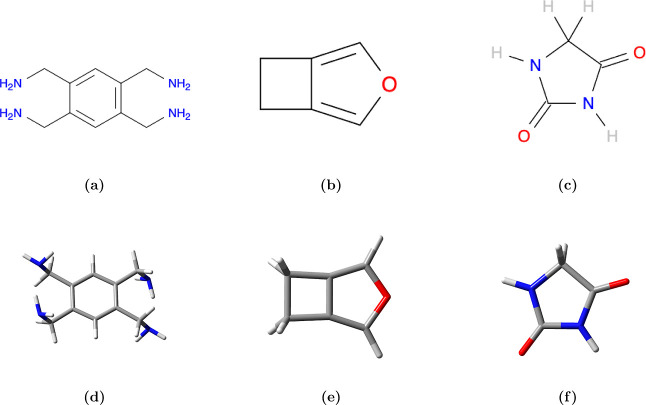
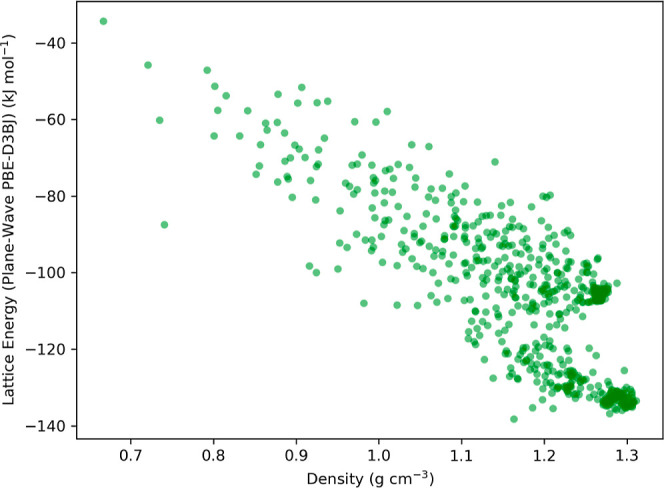
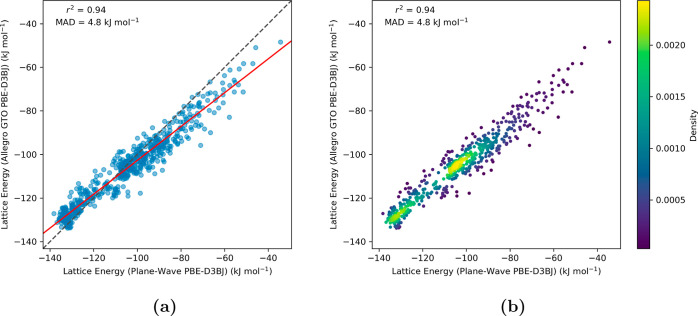
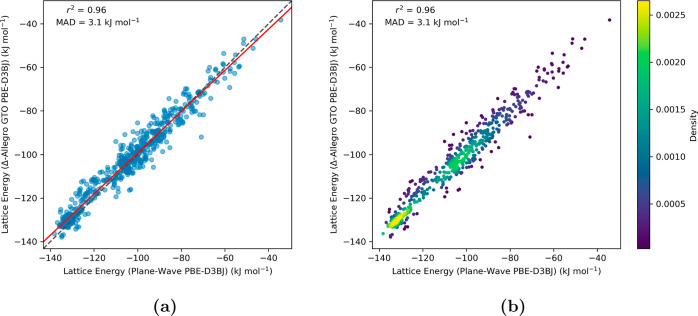
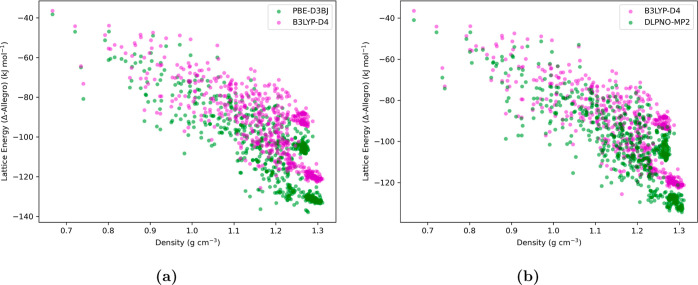
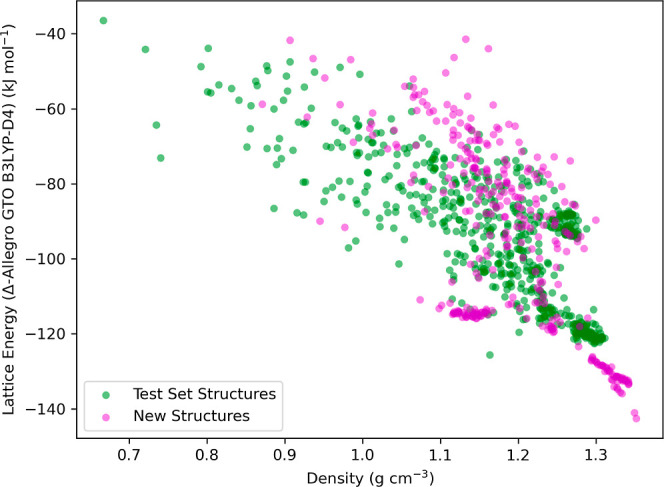
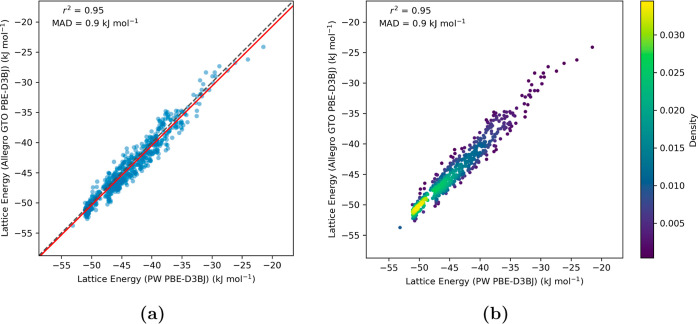
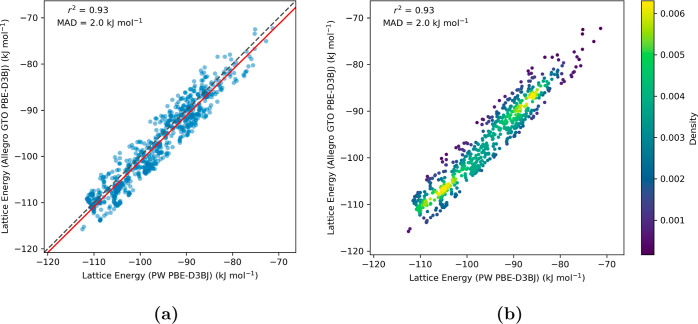
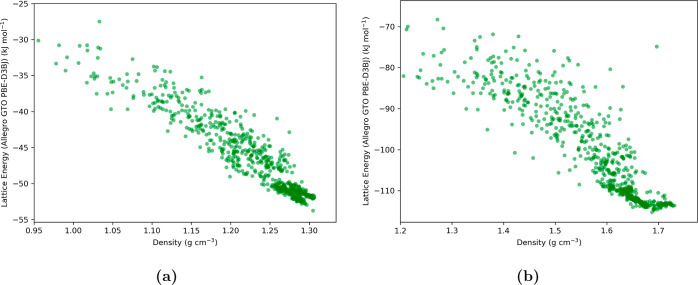
Equivariant neural networks have emerged as prominent models in advancing the construction of interatomic potentials due to their remarkable data efficiency and generalization capabilities for out-of-distribution data. Here, we expand the utility of these networks to the prediction of crystal structures consisting of organic molecules. Traditional methods for computing crystal structure properties, such as plane-wave quantum chemical methods based on density functional theory (DFT), are prohibitively resource-intensive, often necessitating compromises in accuracy and the choice of exchange-correlation functional. We present an approach that leverages the efficiency, and transferability of equivariant neural networks, specifically Allegro, to predict molecular crystal structure energies at a reduced computational cost. Our neural network is trained on molecular clusters using a highly accurate Gaussian-type orbital (GTO)-based method as the target level of theory, eliminating the need for costly periodic DFT calculations, while providing access to all families of exchange-corelation functionals and post-Hartree-Fock methods. The trained model exhibits remarkable accuracy in predicting lattice energies, aligning closely with those computed by plane-wave based DFT methods, thus representing significant cost reductions. Furthermore, the Allegro network was seamlessly integrated with the USPEX framework, accelerating the discovery of low-energy crystal structures during crystal structure prediction.
等变神经网络因其卓越的数据效率和对分布外数据的泛化能力,已成为推进原子间势构建的重要模型。在此,我们将这些网络的效用扩展至由有机分子组成的晶体结构预测。计算晶体结构性质的传统方法,如基于密度泛函理论(DFT)的平面波量子化学方法,资源消耗极大,常常需要在准确性以及交换关联泛函的选择上做出妥协。我们提出一种方法,利用等变神经网络(特别是Allegro)的效率和可转移性,以降低的计算成本预测分子晶体结构能量。我们的神经网络使用基于高精度高斯型轨道(GTO)的方法作为理论目标水平,在分子簇上进行训练,无需进行昂贵的周期性DFT计算,同时可使用所有交换关联泛函族和后哈特里 - 福克方法。训练后的模型在预测晶格能量方面表现出卓越的准确性,与基于平面波的DFT方法计算的结果紧密吻合,从而显著降低了成本。此外,Allegro网络与USPEX框架无缝集成,在晶体结构预测过程中加速了低能量晶体结构的发现。