Department of Computer Engineering, Bilkent University, Ankara, 06800, Turkey.
Tri-Institutional Computational Biology & Medicine Program, Cornell University, 1300 York Ave, New York, 10065, NY, USA.
Genome Biol. 2020 Mar 19;21(1):72. doi: 10.1186/s13059-020-01975-8.
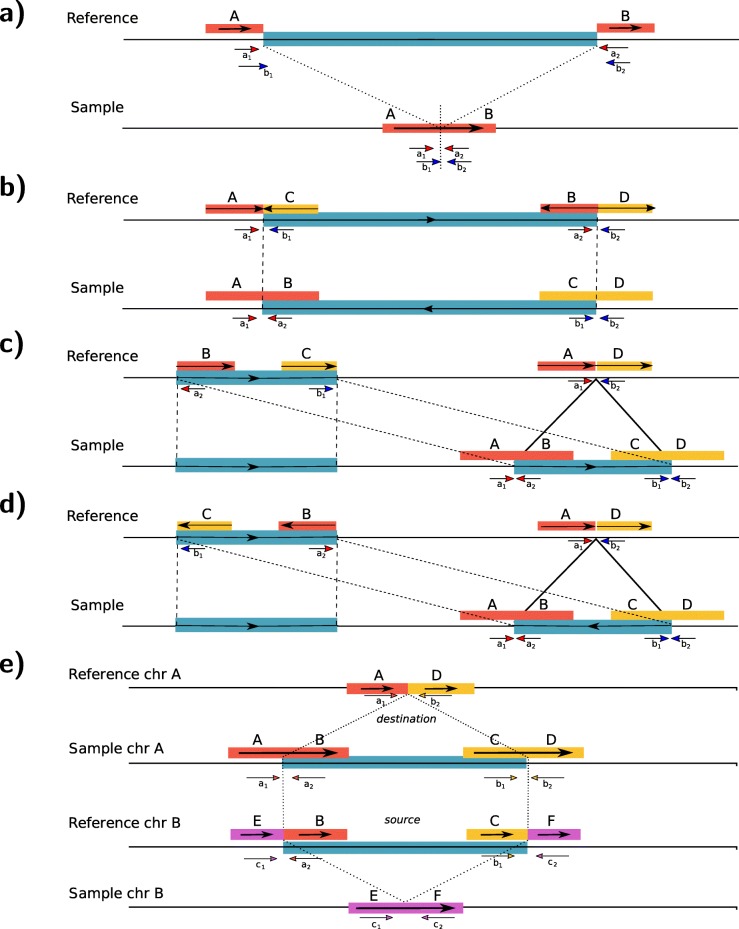
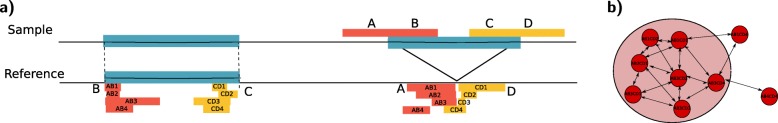
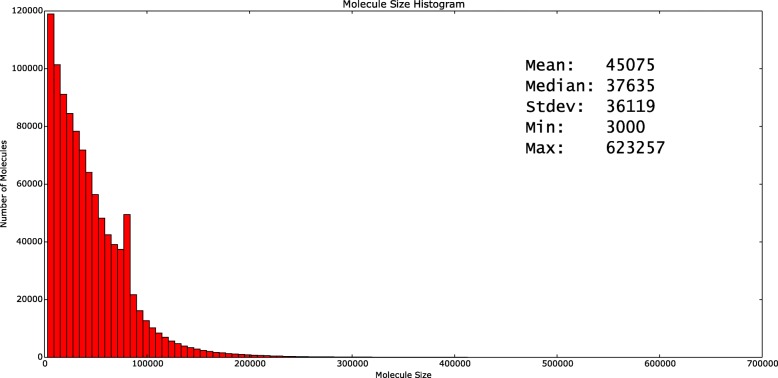
Most existing methods for structural variant detection focus on discovery and genotyping of deletions, insertions, and mobile elements. Detection of balanced structural variants with no gain or loss of genomic segments, for example, inversions and translocations, is a particularly challenging task. Furthermore, there are very few algorithms to predict the insertion locus of large interspersed segmental duplications and characterize translocations. Here, we propose novel algorithms to characterize large interspersed segmental duplications, inversions, deletions, and translocations using linked-read sequencing data. We redesign our earlier algorithm, VALOR, and implement our new algorithms in a new software package, called VALOR2.
大多数现有的结构变异检测方法都侧重于缺失、插入和移动元件的发现和基因分型。例如,平衡结构变异(没有基因组片段的增益或损失)的检测,如倒位和易位,是一项特别具有挑战性的任务。此外,几乎没有算法可以预测大型分散的片段重复插入的位置并对易位进行特征描述。在这里,我们提出了使用链接读取测序数据来对大型分散的片段重复、倒位、缺失和易位进行特征描述的新算法。我们重新设计了早期的算法 VALOR,并在一个名为 VALOR2 的新软件包中实现了我们的新算法。