Division of Neurodegenerative Disorders, St Boniface Hospital Albrechtsen Research Centre, R4046 - 351 Taché Ave, Winnipeg, Manitoba, R2H 2A6, Canada.
Department of Pharmacology and Therapeutics, University of Manitoba, Winnipeg, MB, Canada.
Mol Neurobiol. 2020 Jun;57(6):2521-2538. doi: 10.1007/s12035-020-01900-x. Epub 2020 Mar 20.
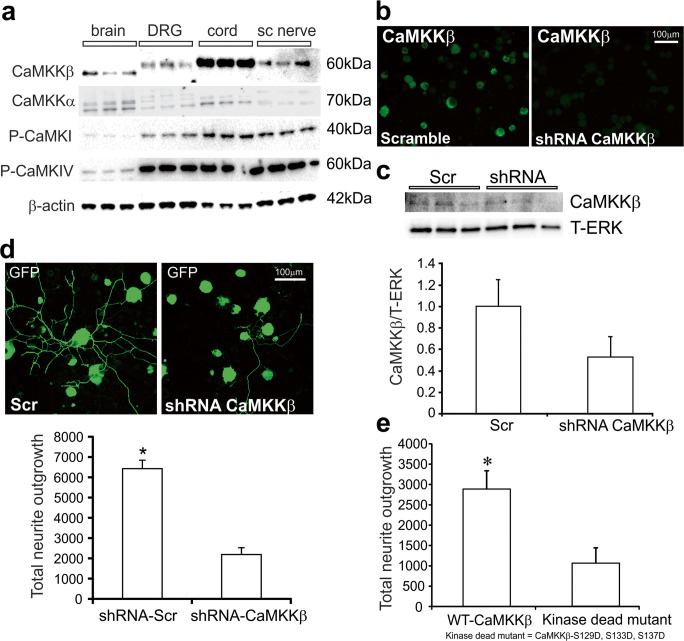
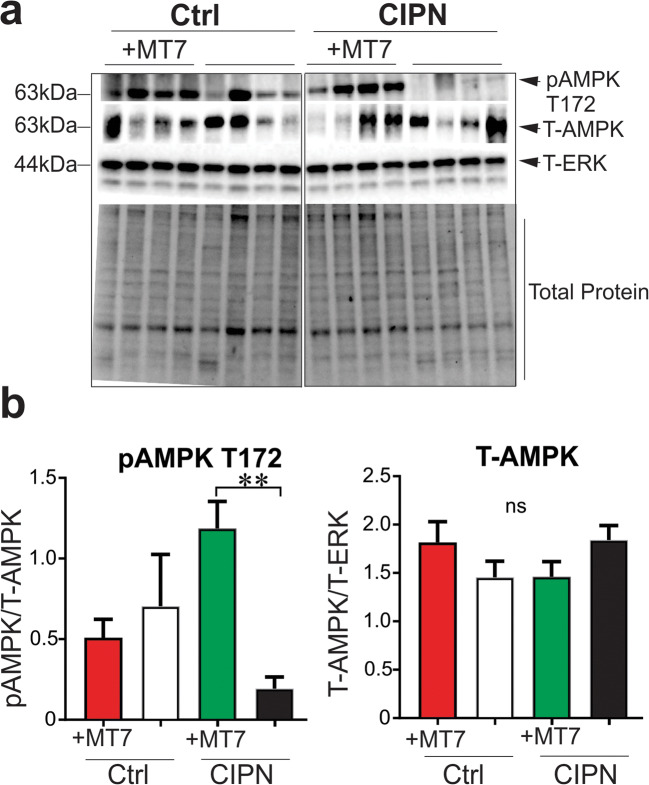
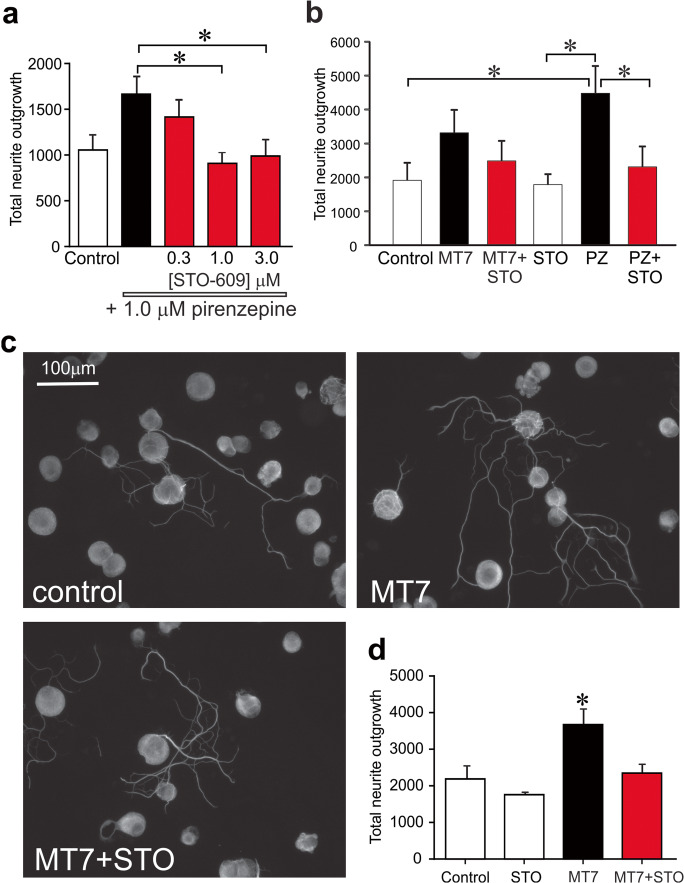
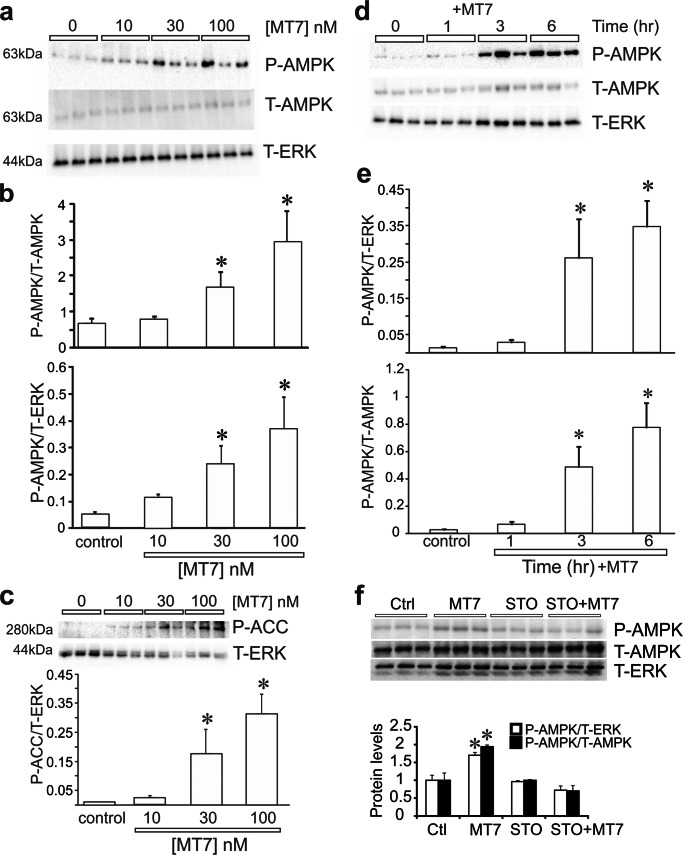
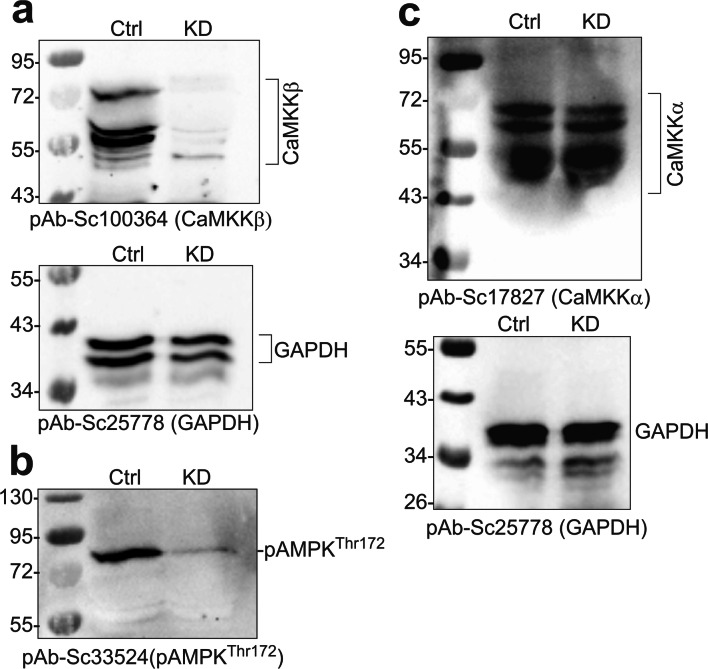
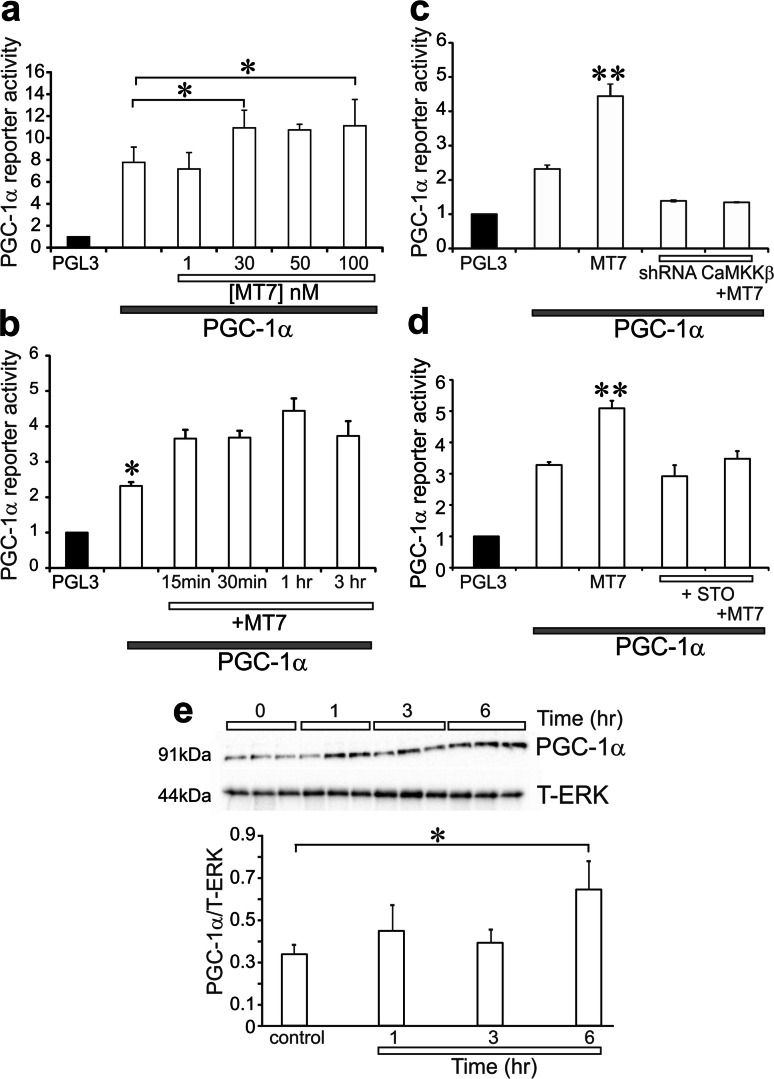
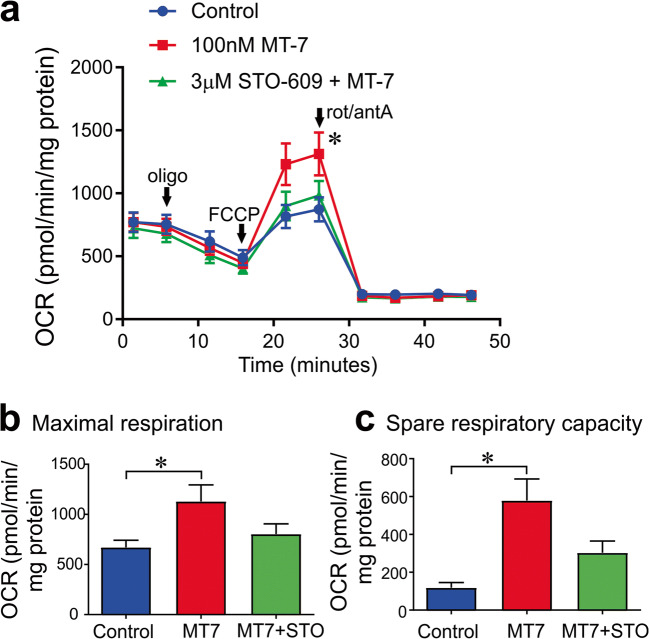
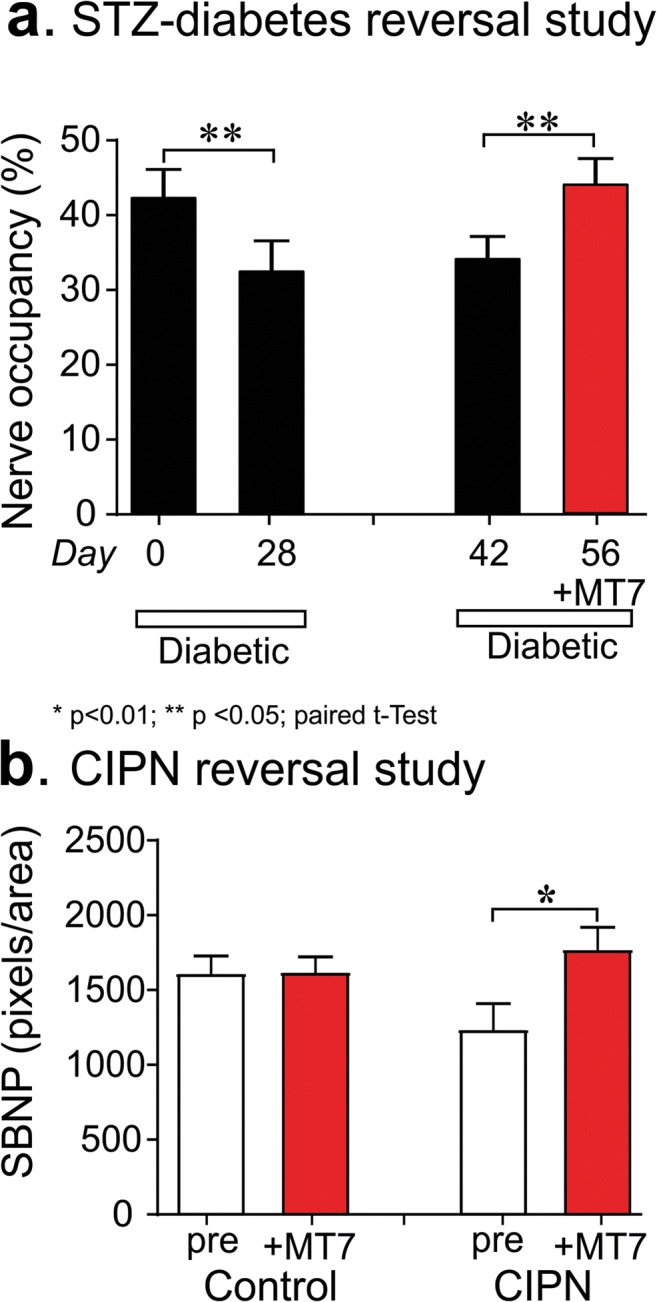
Mitochondrial dysfunction is implicated in a variety of neurodegenerative diseases of the nervous system. Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is a regulator of mitochondrial function in multiple cell types. In sensory neurons, AMP-activated protein kinase (AMPK) augments PGC-1α activity and this pathway is depressed in diabetes leading to mitochondrial dysfunction and neurodegeneration. Antimuscarinic drugs targeting the muscarinic acetylcholine type 1 receptor (MR) prevent/reverse neurodegeneration by inducing nerve regeneration in rodent models of diabetes and chemotherapy-induced peripheral neuropathy (CIPN). Ca/calmodulin-dependent protein kinase kinase β (CaMKKβ) is an upstream regulator of AMPK activity. We hypothesized that antimuscarinic drugs modulate CaMKKβ to enhance activity of AMPK, and PGC-1α, increase mitochondrial function and thus protect from neurodegeneration. We used the specific MR antagonist muscarinic toxin 7 (MT7) to manipulate muscarinic signaling in the dorsal root ganglia (DRG) neurons of normal rats or rats with streptozotocin-induced diabetes. DRG neurons treated with MT7 (100 nM) or a selective muscarinic antagonist, pirenzepine (1 μM), for 24 h showed increased neurite outgrowth that was blocked by the CaMKK inhibitor STO-609 (1 μM) or short hairpin RNA to CaMKKβ. MT7 enhanced AMPK phosphorylation which was blocked by STO-609 (1 μM). PGC-1α reporter activity was augmented up to 2-fold (p < 0.05) by MT7 and blocked by STO-609. Mitochondrial maximal respiration and spare respiratory capacity were elevated after 3 h of exposure to MT7 (p < 0.05). Diabetes and CIPN induced a significant (p < 0.05) decrease in corneal nerve density which was corrected by topical delivery of MT7. We reveal a novel MR-modulated, CaMKKβ-dependent pathway in neurons that represents a therapeutic target to enhance nerve repair in two of the most common forms of peripheral neuropathy.
线粒体功能障碍与多种神经系统退行性疾病有关。过氧化物酶体增殖物激活受体γ共激活因子 1α(PGC-1α)是多种细胞类型中线粒体功能的调节剂。在感觉神经元中,AMP 激活的蛋白激酶(AMPK)增强 PGC-1α 的活性,而在糖尿病中,该途径被抑制,导致线粒体功能障碍和神经退行性变。针对毒蕈碱乙酰胆碱 M1 型受体(MR)的抗毒蕈碱药物通过在糖尿病和化疗诱导的周围神经病变(CIPN)的啮齿动物模型中诱导神经再生来预防/逆转神经退行性变。钙/钙调蛋白依赖性蛋白激酶激酶β(CaMKKβ)是 AMPK 活性的上游调节剂。我们假设抗毒蕈碱药物调节 CaMKKβ 以增强 AMPK 和 PGC-1α 的活性,增加线粒体功能,从而防止神经退行性变。我们使用特异性 MR 拮抗剂毒蕈碱毒素 7(MT7)在正常大鼠或链脲佐菌素诱导的糖尿病大鼠的背根神经节(DRG)神经元中操纵毒蕈碱信号。用 MT7(100 nM)或选择性毒蕈碱拮抗剂哌仑西平(1 μM)处理 24 小时的 DRG 神经元显示出突起生长增加,该增加被 CaMKK 抑制剂 STO-609(1 μM)或 CaMKKβ的短发夹 RNA 阻断。MT7 增强 AMPK 磷酸化,该磷酸化被 STO-609(1 μM)阻断。MT7 将 PGC-1α 报告基因活性增加了 2 倍(p < 0.05),并被 STO-609 阻断。暴露于 MT7 3 小时后,线粒体最大呼吸和备用呼吸能力升高(p < 0.05)。糖尿病和 CIPN 诱导角膜神经密度显著降低(p < 0.05),这通过局部给予 MT7 得到纠正。我们揭示了一种新的神经元中 MR 调节的 CaMKKβ依赖性途径,它代表了增强两种最常见的周围神经病变中神经修复的治疗靶点。