Research Center of Combined Traditional Chinese and Western Medicine, Affiliated Traditional Medicine Hospital, Southwest Medical University, Luzhou, Sichuan, China (mainland).
National Traditional Chinese Medicine Clinical Research Base, Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, Sichuan, China (mainland).
Med Sci Monit. 2020 Mar 22;26:e920854. doi: 10.12659/MSM.920854.
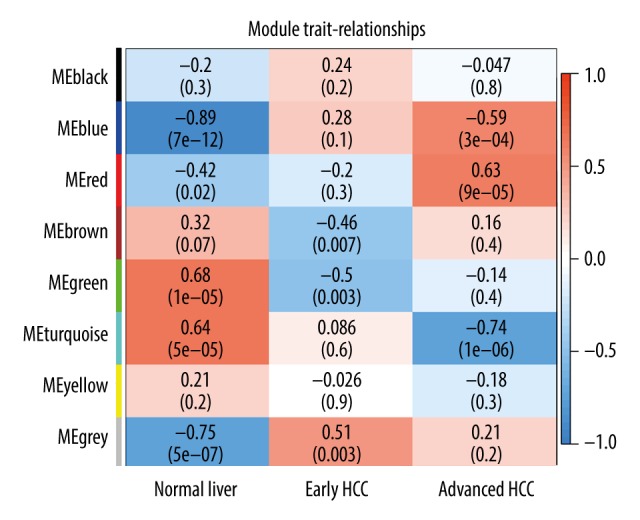
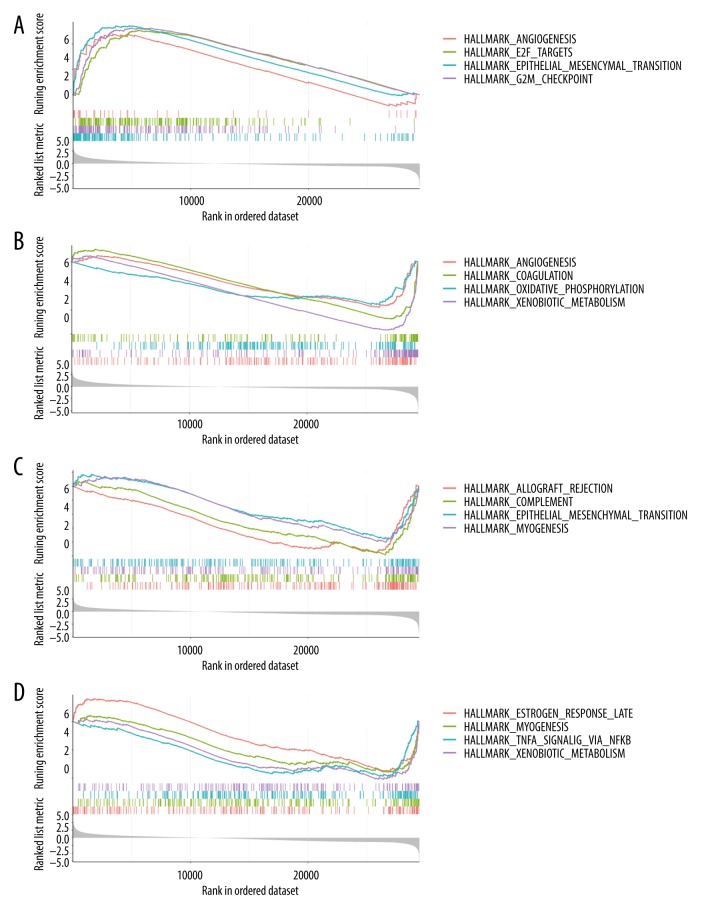
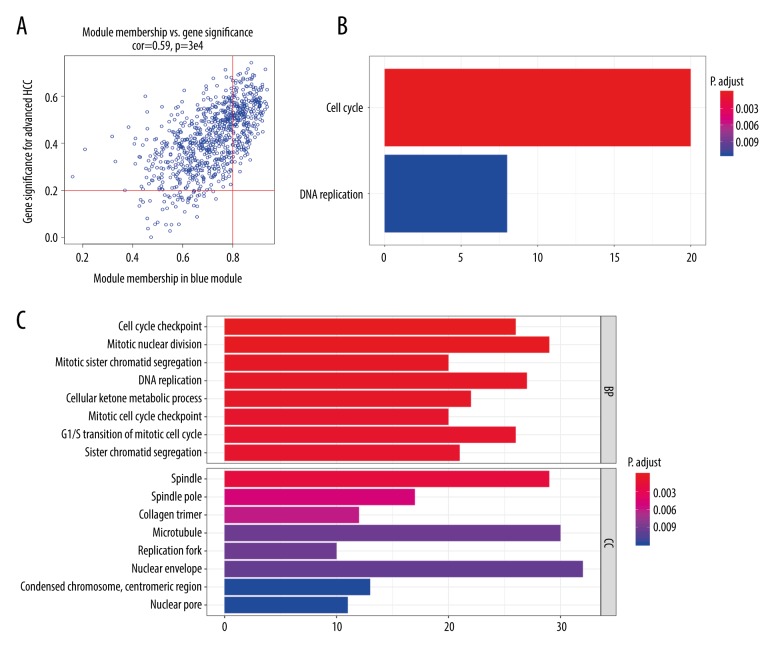
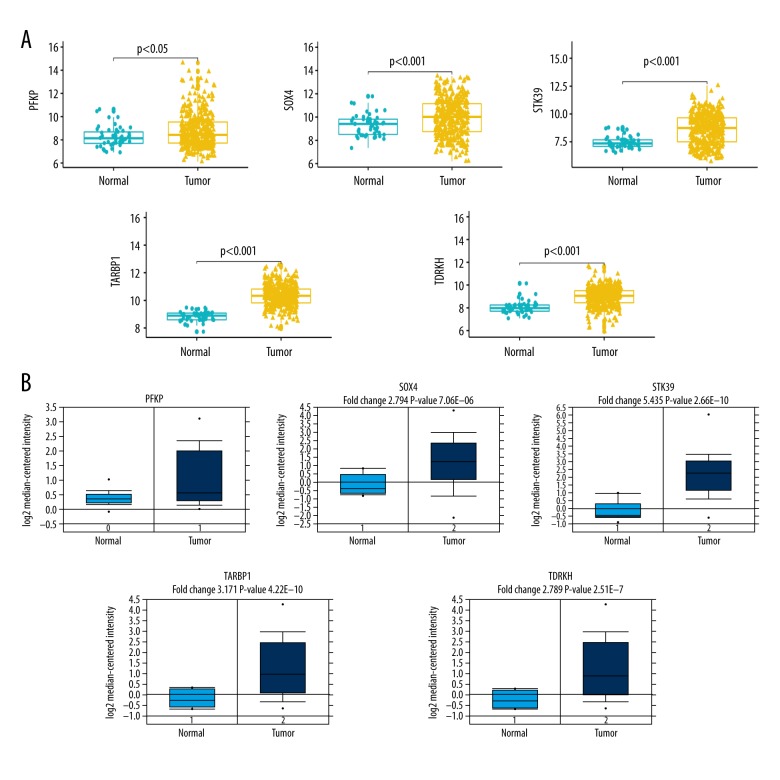
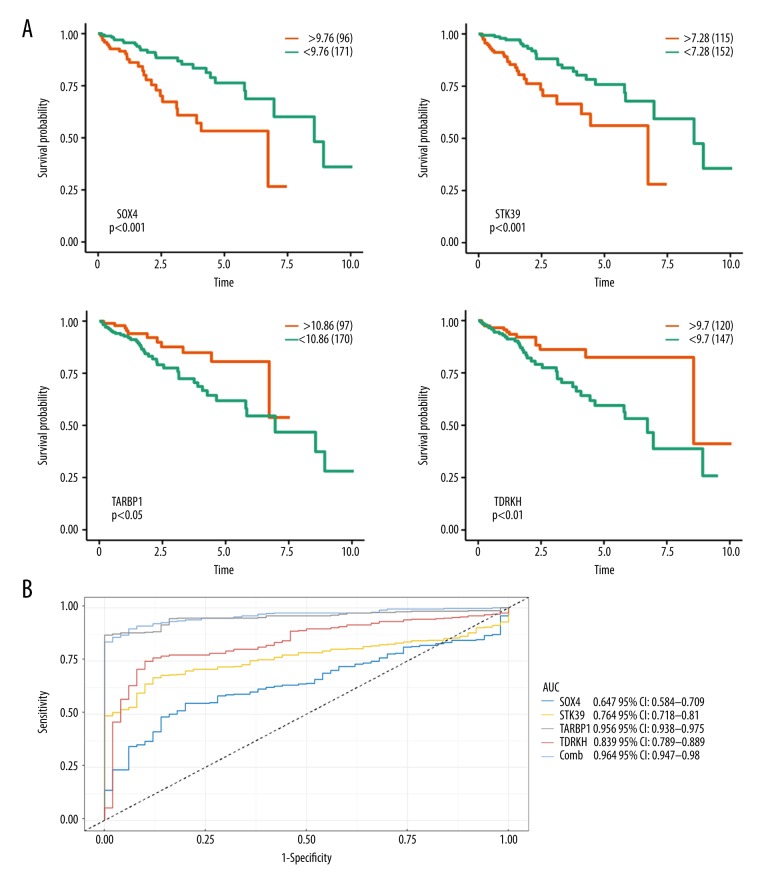
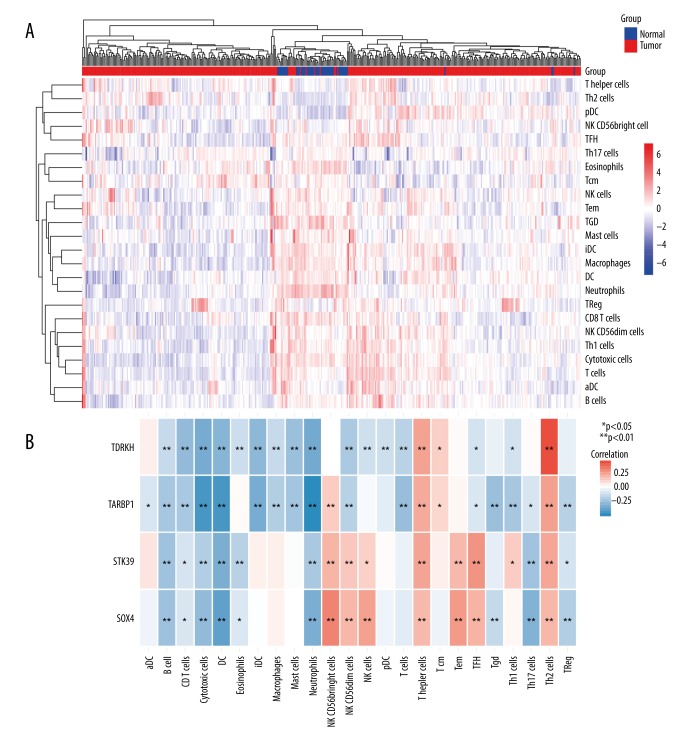
BACKGROUND Hepatocellular carcinoma (HCC) is one of the most prevalent cancers in the world. Bioinformatics studies have been widely used for screening genes involved in the initiation and progression of HCC. MATERIAL AND METHODS We obtained liver cancer microarray raw data from the GEO database (GSE54238). Next, weighted gene co-expression network analysis (WGCNA) was used to assess the critical modules. Then, we assessed the gene significance by calculating survival, expression level, and receiver operating characteristic (ROC) in the TCGA database. We also validated the expression of selected genes in the Oncomine database and calculated the relationship between 4 hub genes and immune infiltration. Finally, GSEA enrichment analysis was used to explore the potential mechanism. RESULTS We identified the red and blue modules as the critical modules, and found 176 candidate genes by assessing gene significance. GO and KEEG results suggested that the candidate genes are involved in the cell cycle. Four hub genes - SOX4, STK39, TARBP1, and TDRKH - were eventually screened after validating their expression and power in diagnosing HCC in the TCGA database. Immune infiltration analysis and GSEA enrichment analysis showed that these 4 hub genes were correlated with the immune cell populations infiltration and that multiple mechanisms were involved, such as angiogenesis and epithelial-mesenchymal transition. CONCLUSIONS Our findings revealed that these 4 genes can be regarded as potential prognosticators and therapeutic targets for HCC.
肝细胞癌(HCC)是世界上最常见的癌症之一。生物信息学研究已广泛用于筛选与 HCC 发生和发展相关的基因。
我们从 GEO 数据库(GSE54238)中获取肝癌微阵列原始数据。接下来,使用加权基因共表达网络分析(WGCNA)来评估关键模块。然后,我们通过计算 TCGA 数据库中的生存、表达水平和接受者操作特征(ROC)来评估基因的重要性。我们还在 Oncomine 数据库中验证了选定基因的表达,并计算了 4 个枢纽基因与免疫浸润之间的关系。最后,使用 GSEA 富集分析来探索潜在的机制。
我们确定红色和蓝色模块为关键模块,并通过评估基因的重要性发现了 176 个候选基因。GO 和 KEEG 结果表明,候选基因参与细胞周期。经过在 TCGA 数据库中验证其在诊断 HCC 中的表达和效能,最终筛选出 4 个枢纽基因-SOX4、STK39、TARBP1 和 TDRKH。免疫浸润分析和 GSEA 富集分析表明,这 4 个枢纽基因与免疫细胞群浸润相关,涉及多种机制,如血管生成和上皮-间充质转化。
我们的研究结果表明,这 4 个基因可以作为 HCC 的潜在预后标志物和治疗靶点。