Department of Medicine, University of Cambridge, Box 157 Addenbrooke's Hospital, Hills Road, Cambridge, CB2 0QQ, UK.
Next Gen Diagnostics LLC (NGD), Mountain View, CA and Wellcome Genome Campus, Hinxton, Cambridge, UK.
Microb Genom. 2020 Apr;6(4). doi: 10.1099/mgen.0.000354. Epub 2020 Mar 31.
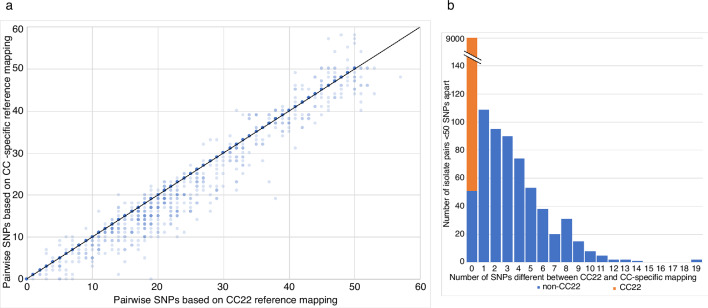
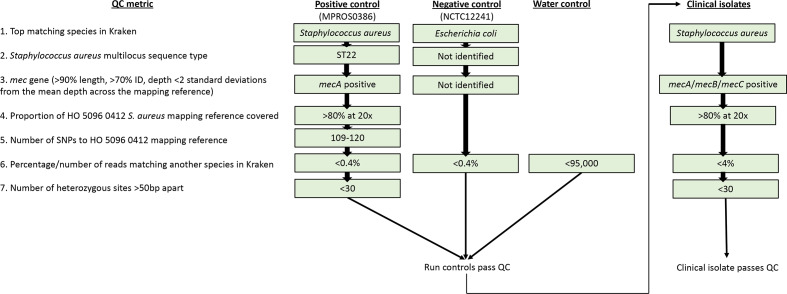
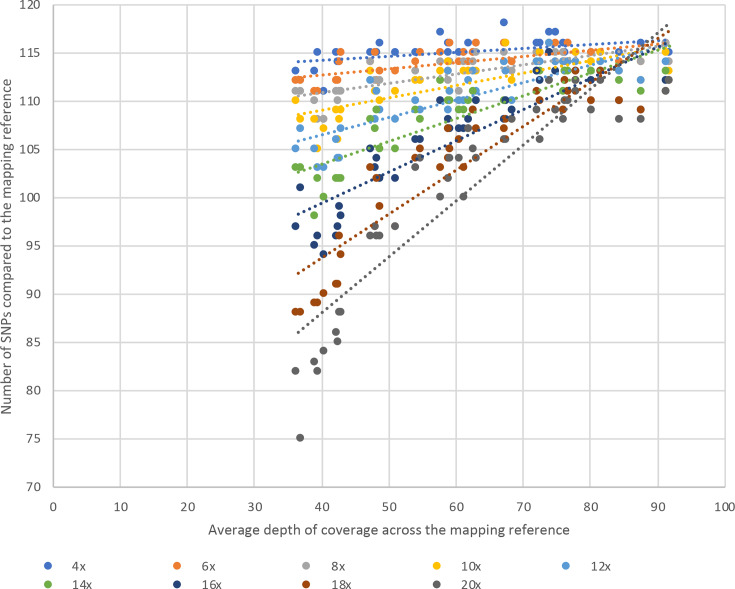
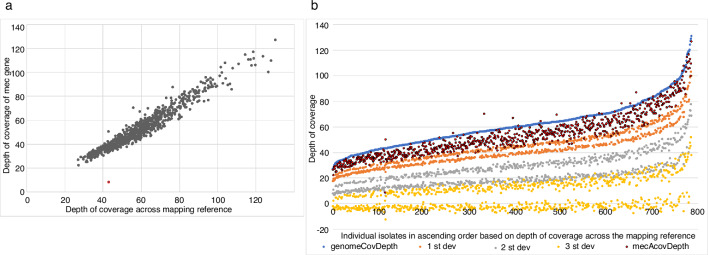
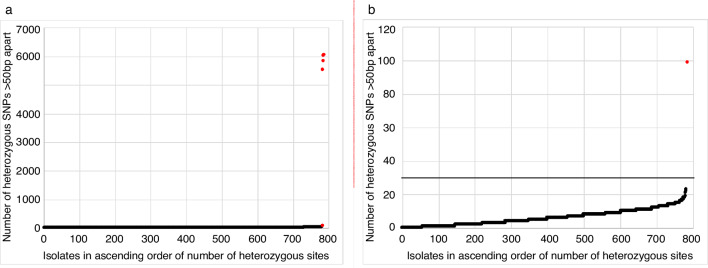
Bacterial sequencing will become increasingly adopted in routine microbiology laboratories. Here, we report the findings of a technical evaluation of almost 800 clinical methicillin-resistant (MRSA) isolates, in which we sought to define key quality metrics to support MRSA sequencing in clinical practice. We evaluated the accuracy of mapping to a generic reference versus clonal complex (CC)-specific mapping, which is more computationally challenging. Focusing on isolates that were genetically related (<50 single nucleotide polymorphisms (SNPs)) and belonged to prevalent sequence types, concordance between these methods was 99.5 %. We use MRSA MPROS0386 to control for base calling accuracy by the sequencer, and used multiple repeat sequences of the control to define a permitted range of SNPs different to the mapping reference for this control (equating to 3 standard deviations from the mean). Repeat sequences of the control were also used to demonstrate that SNP calling was most accurate across differing coverage depths (above 35×, the lowest depth in our study) when the depth required to call a SNP as present was at least 4-8×. Using 786 MRSA sequences, we defined a robust measure for gene detection to reduce false-positives arising from contamination, which was no greater than 2 standard deviations below the average depth of coverage across the genome. Sequencing from bacteria harvested from clinical plates runs an increased risk of contamination with the same or different species, and we defined a cut-off of 30 heterozygous sites >50 bp apart to identify same-species contamination for MRSA. These metrics were combined into a quality-control (QC) flowchart to determine whether sequence runs and individual clinical isolates passed QC, which could be adapted by future automated analysis systems to enable rapid hands-off sequence analysis by clinical laboratories.
细菌测序将越来越多地被常规微生物学实验室采用。在这里,我们报告了近 800 株临床耐甲氧西林金黄色葡萄球菌 (MRSA) 分离株的技术评估结果,我们试图确定关键的质量指标,以支持 MRSA 测序在临床实践中的应用。我们评估了映射到通用参考与克隆复合体 (CC)-特异性映射的准确性,后者更具计算挑战性。我们专注于遗传上相关的 (<50 个单核苷酸多态性 (SNP)) 且属于常见序列型的分离株,这两种方法之间的一致性为 99.5%。我们使用 MRSA MPROS0386 来控制测序仪的碱基调用准确性,并使用该对照物的多个重复序列来定义一个允许的 SNP 范围,与该对照物的映射参考不同 (相当于平均值的 3 个标准差)。我们还使用对照物的重复序列来证明,当需要调用 SNP 存在的深度至少为 4-8 倍时,SNP 调用在不同的覆盖深度 (我们研究中最低的深度为 35 倍以上) 下最为准确。使用 786 株 MRSA 序列,我们定义了一种可靠的基因检测方法,以减少由于污染而产生的假阳性,其值不超过基因组覆盖的平均深度的 2 个标准差。从临床平板上收获的细菌进行测序会增加与相同或不同物种污染的风险,我们定义了一个 30 个异质位点 (>50 bp 间隔) 的截止值,以识别 MRSA 的同种污染。这些指标被组合到一个质量控制 (QC) 流程图中,以确定序列运行和单个临床分离株是否通过 QC,未来的自动化分析系统可以采用这些指标,使临床实验室能够快速进行无人值守的序列分析。