Naorem Romen Singh, Urban Peter, Goswami Gunajit, Fekete Csaba
Department of General and Environmental Microbiology, Institute of Biology, University of Pécs, Pécs, 7624 Hungary.
Microbial Biotechnology Research Group, Szentágothai Research Centre, Pécs, 7624 Hungary.
3 Biotech. 2020 Sep;10(9):401. doi: 10.1007/s13205-020-02387-y. Epub 2020 Aug 20.
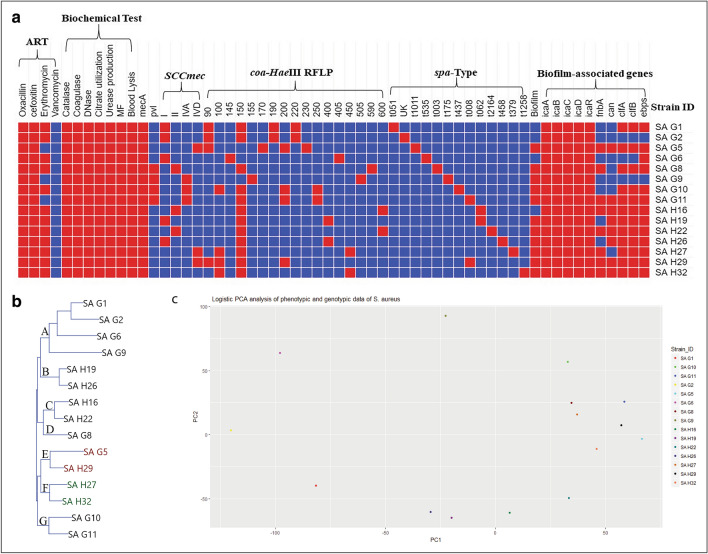
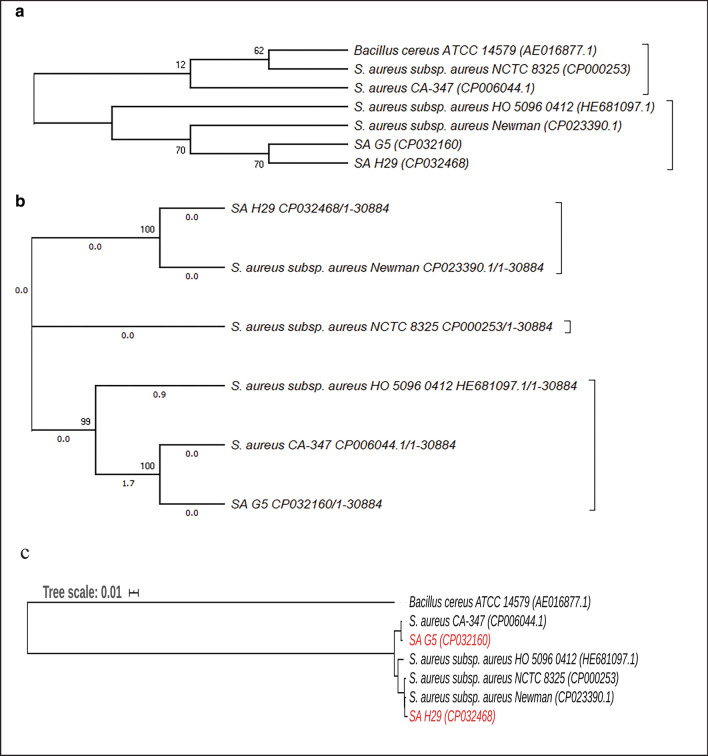
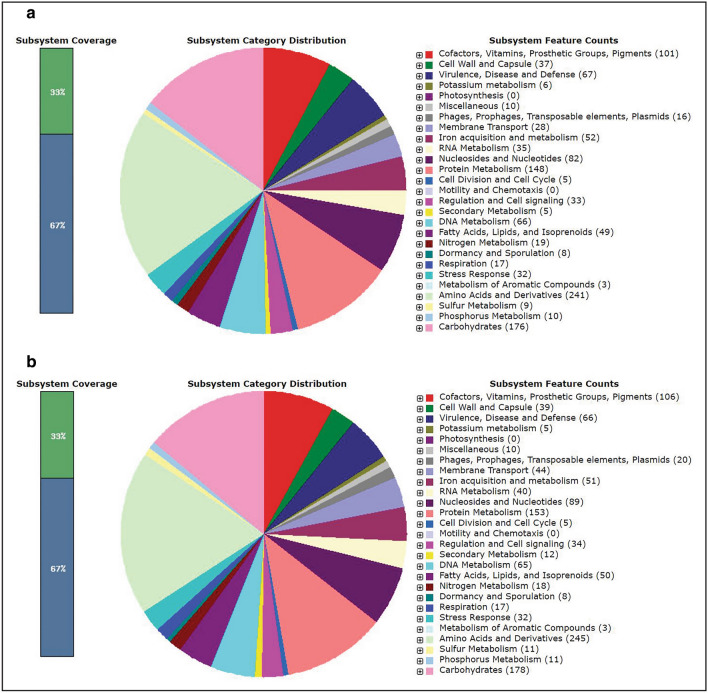
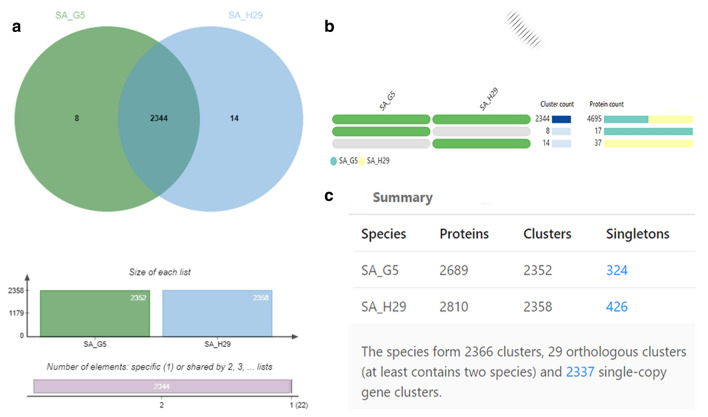
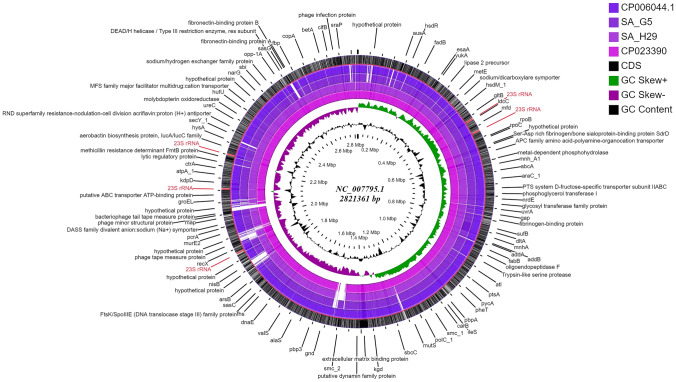
In the present study, a total of 35 isolates collected from two different geographical locations viz., Germany and Hungary were tested for their methicillin-resistant phenotype which revealed a high incidence of methicillin-resistant . The quantitative test for biofilm production revealed that 73.3% of isolates were biofilm producers. The isolates were further characterized using a set of biochemical and genotypic methods such as amplification and analysis of species-specific sequence and gene. The 33 positive isolates were then characterized by the amplification of and toxin genes. Further, based on the biofilm-forming phenotype, 15 isolates were selected and characterized through PCR-RFLP of gene, polymorphism of gene and amplification of biofilm-associated genes. The dendrogram prepared from the results of both biochemical and genotypic analyses of the 15 isolates showed that except for the isolates SA G5 and SA H29, the rest of the isolates grouped themselves according to their locations. Thus, the two isolates were selected for further characterization through whole-genome sequencing. Comparative genome analysis revealed that SA G5 and SA H29 have 97.20% ANI values with 2344 gene clusters (core-genome) of which 16 genes were related to antibiotic resistance genes and 57 genes encode virulence factors. The highest numbers of singleton genes were found in SA H29 that encodes proteins for virulence, resistance, mobile elements, and lanthionine biosynthesis. The high-resolution phylogenetic trees generated based on shared proteins and SNPs revealed a clear difference between the two strains and can be useful in distinguishing closely related genomes. The present study demonstrated that the whole-genome sequence analysis technique is required to get a better insight into the MRSA strains which would be helpful in improving diagnostic investigations in real-time to improve patient care.
在本研究中,对从两个不同地理位置(即德国和匈牙利)收集的总共35株分离株进行了耐甲氧西林表型测试,结果显示耐甲氧西林的发生率很高。生物膜产生的定量测试表明,73.3%的分离株是生物膜产生菌。使用一组生化和基因分型方法对分离株进行了进一步表征,例如物种特异性序列和基因的扩增及分析。然后通过扩增和毒素基因对33株阳性分离株进行了表征。此外,基于生物膜形成表型,选择了15株分离株,并通过基因的PCR-RFLP、基因的多态性和生物膜相关基因的扩增进行了表征。根据15株分离株的生化和基因分析结果绘制的系统发育树表明,除了分离株SA G5和SA H29外,其余分离株根据其地理位置进行了分组。因此,选择这两株分离株通过全基因组测序进行进一步表征。比较基因组分析显示,SA G5和SA H29的ANI值为97.20%,有2344个基因簇(核心基因组),其中16个基因与抗生素抗性基因相关,57个基因编码毒力因子。在SA H29中发现的单基因数量最多,这些基因编码毒力、抗性、移动元件和羊毛硫氨酸生物合成的蛋白质。基于共享蛋白质和SNP生成的高分辨率系统发育树显示这两个菌株之间存在明显差异,可用于区分密切相关基因组。本研究表明,需要全基因组序列分析技术来更好地了解耐甲氧西林金黄色葡萄球菌菌株,这将有助于实时改进诊断研究以改善患者护理。