Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, Worcester, Massachusetts, USA.
Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, Worcester, Massachusetts, USA
mBio. 2020 Mar 31;11(2):e00172-20. doi: 10.1128/mBio.00172-20.
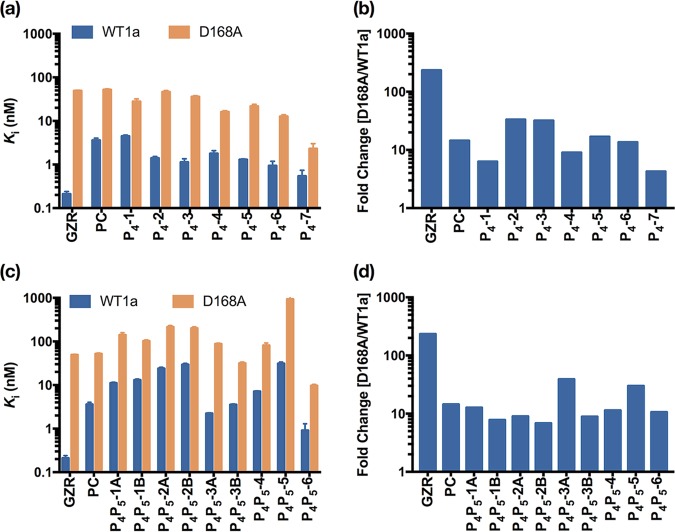
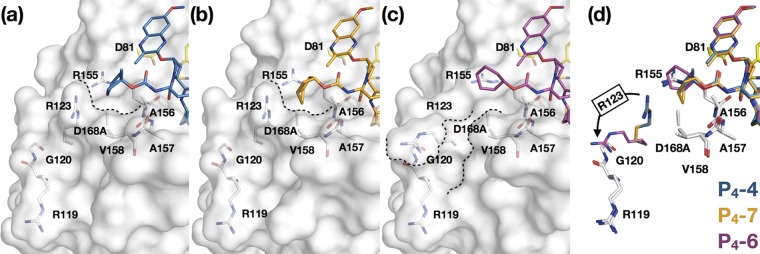
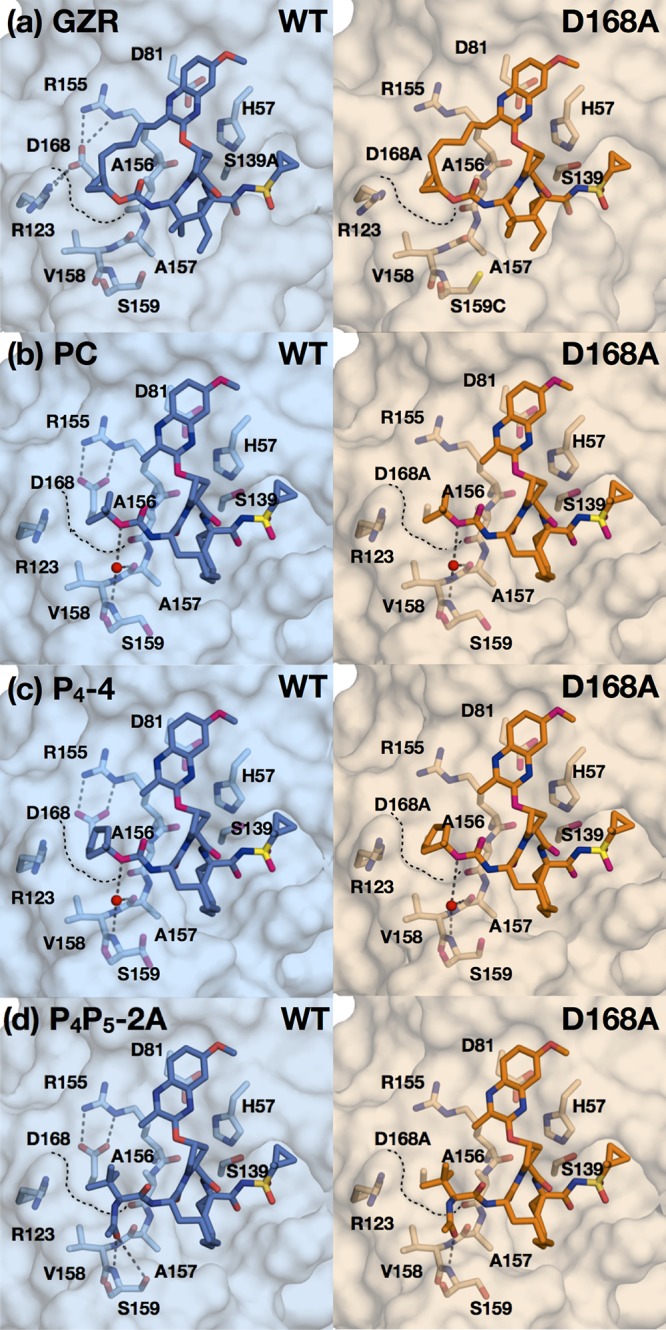
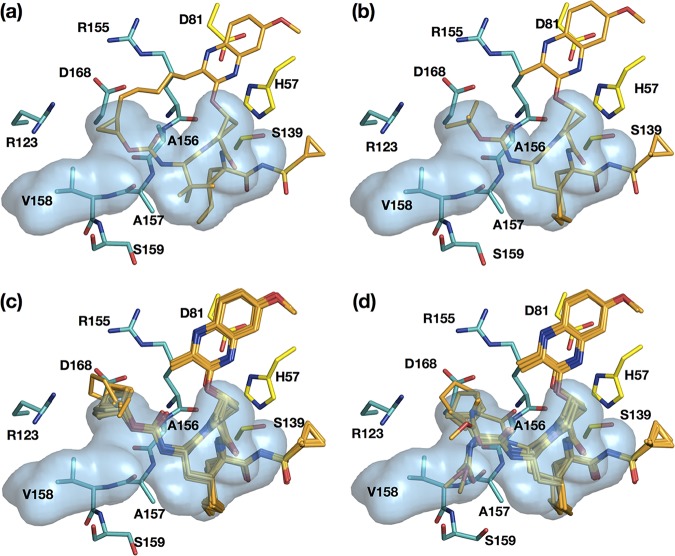
Hepatitis C virus (HCV) infects millions of people worldwide, causing chronic liver disease that can lead to cirrhosis, hepatocellular carcinoma, and liver transplant. In the last several years, the advent of direct-acting antivirals, including NS3/4A protease inhibitors (PIs), has remarkably improved treatment outcomes of HCV-infected patients. However, selection of resistance-associated substitutions and polymorphisms among genotypes can lead to drug resistance and in some cases treatment failure. A proactive strategy to combat resistance is to constrain PIs within evolutionarily conserved regions in the protease active site. Designing PIs using the substrate envelope is a rational strategy to decrease the susceptibility to resistance by using the constraints of substrate recognition. We successfully designed two series of HCV NS3/4A PIs to leverage unexploited areas in the substrate envelope to improve potency, specifically against resistance-associated substitutions at D168. Our design strategy achieved better resistance profiles over both the FDA-approved NS3/4A PI grazoprevir and the parent compound against the clinically relevant D168A substitution. Crystallographic structural analysis and inhibition assays confirmed that optimally filling the substrate envelope is critical to improve inhibitor potency while avoiding resistance. Specifically, inhibitors that enhanced hydrophobic packing in the S4 pocket and avoided an energetically frustrated pocket performed the best. Thus, the HCV substrate envelope proved to be a powerful tool to design robust PIs, offering a strategy that can be translated to other targets for rational design of inhibitors with improved potency and resistance profiles. Despite significant progress, hepatitis C virus (HCV) continues to be a major health problem with millions of people infected worldwide and thousands dying annually due to resulting complications. Recent antiviral combinations can achieve >95% cure, but late diagnosis, low access to treatment, and treatment failure due to drug resistance continue to be roadblocks against eradication of the virus. We report the rational design of two series of HCV NS3/4A protease inhibitors with improved resistance profiles by exploiting evolutionarily constrained regions of the active site using the substrate envelope model. Optimally filling the S4 pocket is critical to avoid resistance and improve potency. Our results provide drug design strategies to avoid resistance that are applicable to other quickly evolving viral drug targets.
丙型肝炎病毒(HCV)感染了全球数百万人,导致慢性肝病,可导致肝硬化、肝细胞癌和肝移植。在过去的几年中,直接作用抗病毒药物(包括 NS3/4A 蛋白酶抑制剂(PI))的出现极大地改善了 HCV 感染患者的治疗效果。然而,基因型中耐药相关取代和多态性的选择可导致耐药性,在某些情况下还会导致治疗失败。对抗耐药性的积极策略是将 PI 限制在蛋白酶活性部位的进化保守区域内。使用底物包络设计 PI 是一种通过利用底物识别的约束来降低耐药性敏感性的合理策略。我们成功设计了两种 HCV NS3/4A PI 系列,利用底物包络中未开发的区域来提高效力,特别是针对耐药相关的 D168 取代。我们的设计策略在针对 FDA 批准的 NS3/4A PI 格拉西洛韦和母体化合物的情况下,对 D168A 取代均实现了更好的耐药谱。晶体结构分析和抑制测定证实,优化底物包络的填充对于提高抑制剂效力而避免耐药性至关重要。具体来说,增强 S4 口袋中疏水性堆积并避免能量受挫口袋的抑制剂表现最佳。因此,HCV 底物包络被证明是设计强大 PI 的有力工具,为其他目标提供了一种策略,可用于合理设计具有改善效力和耐药谱的抑制剂。尽管取得了重大进展,但丙型肝炎病毒(HCV)仍然是一个重大的健康问题,全球有数百万人感染,每年有数千人因由此导致的并发症而死亡。最近的抗病毒联合疗法可实现>95%的治愈率,但由于诊断较晚、治疗机会有限以及耐药导致的治疗失败,仍存在消除该病毒的障碍。我们报告了两种 HCV NS3/4A 蛋白酶抑制剂系列的合理设计,这些抑制剂通过使用底物包络模型利用活性部位的进化约束区域,改善了耐药谱。最佳填充 S4 口袋对于避免耐药性和提高效力至关重要。我们的结果提供了避免耐药性的药物设计策略,适用于其他快速进化的病毒药物靶标。