Paul G. Allen School for Global Animal Health, Washington State University, Pullman, Washington, USA.
Paul G. Allen School for Global Animal Health, Washington State University, Pullman, Washington, USA
mBio. 2020 Mar 31;11(2):e03350-19. doi: 10.1128/mBio.03350-19.
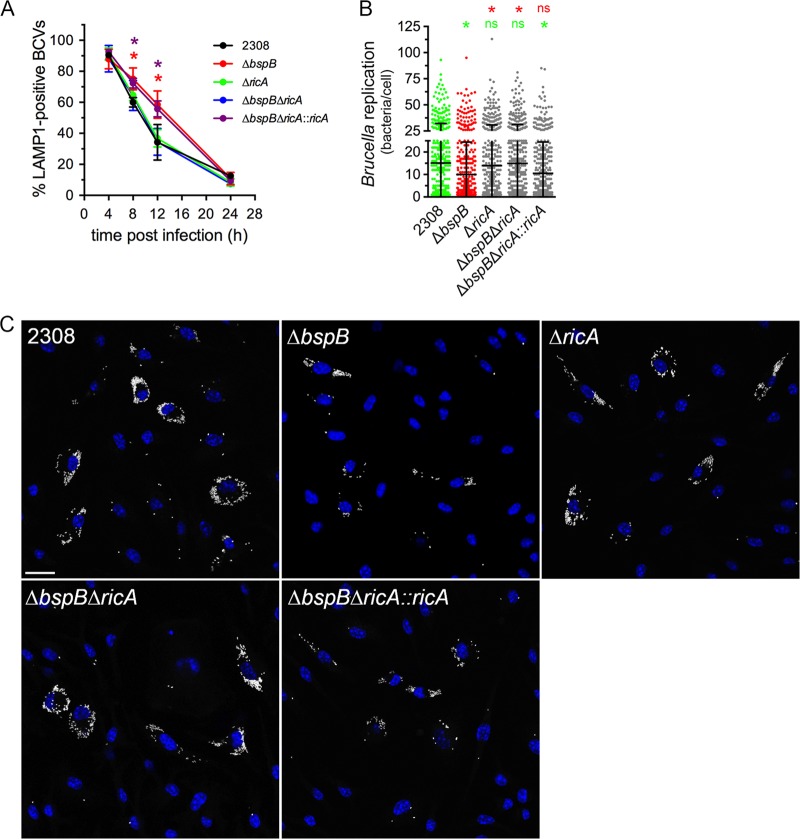
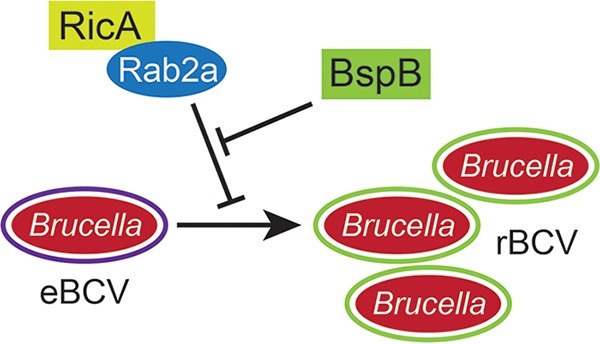
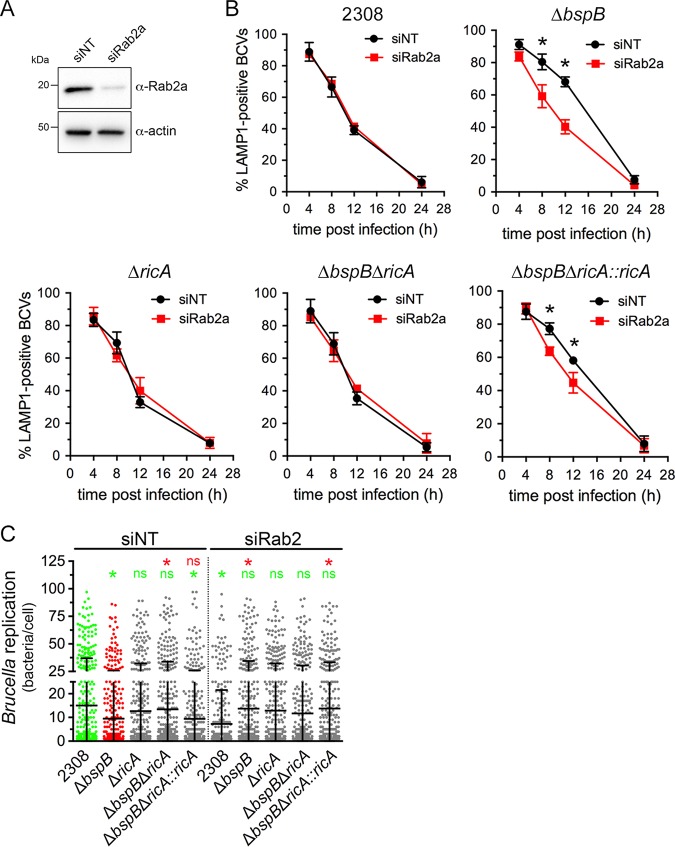
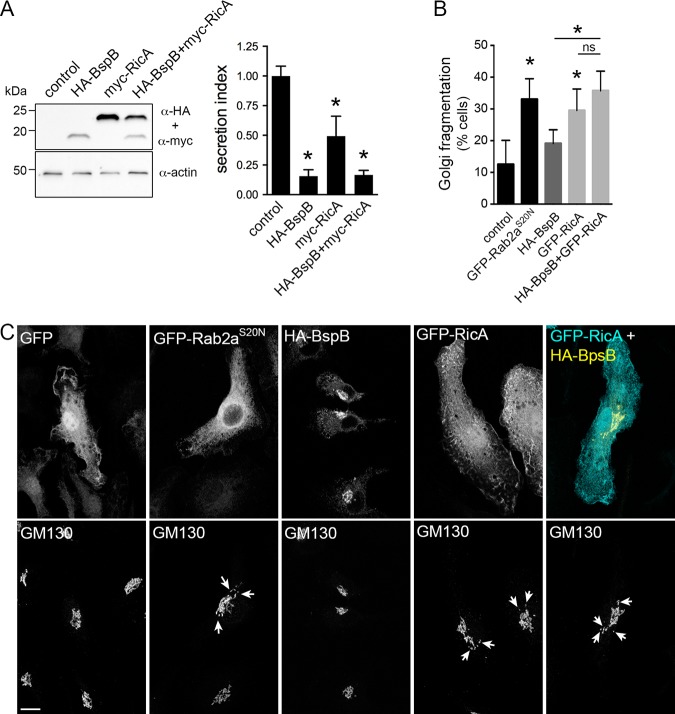
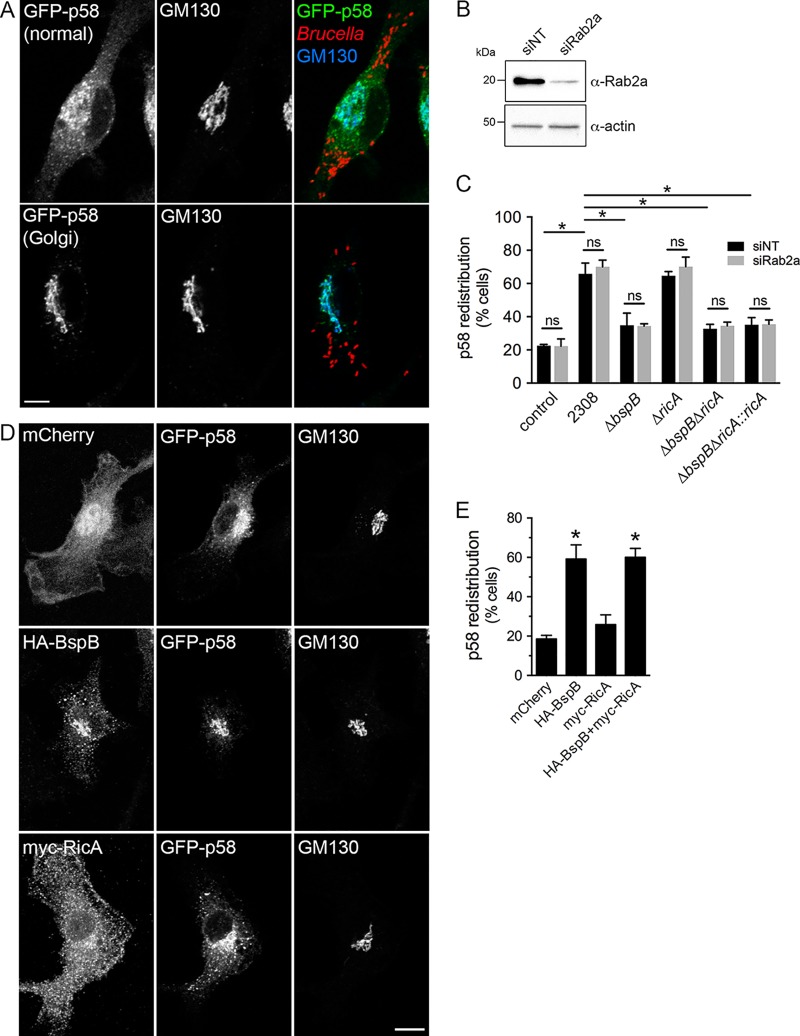
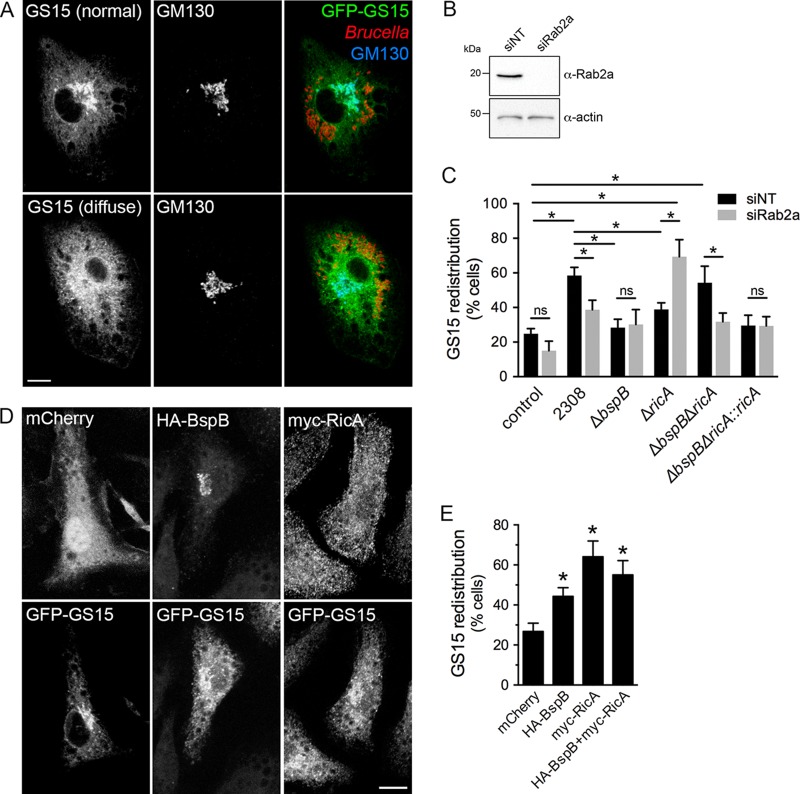
Intracellular bacterial pathogens remodel cellular functions during their infectious cycle via the coordinated actions of effector molecules delivered through dedicated secretion systems. While the function of many individual effectors is known, how they interact to promote pathogenesis is rarely understood. The zoonotic bacterium , the causative agent of brucellosis, delivers effector proteins via its VirB type IV secretion system (T4SS) which mediate biogenesis of the endoplasmic reticulum (ER)-derived replicative -containing vacuole (rBCV). Here, we show that T4SS effectors BspB and RicA display epistatic interactions in replication. Defects in rBCV biogenesis and replication caused by deletion of were dependent on the host GTPase Rab2a and suppressed by the deletion of , indicating a role of Rab2-binding effector RicA in these phenotypic defects. Rab2a requirements for rBCV biogenesis and intracellular replication were abolished upon deletion of both and , demonstrating that the functional interaction of these effectors engages Rab2-dependent transport in the intracellular cycle. Expression of RicA impaired host secretion and caused Golgi fragmentation. While BspB-mediated changes in ER-to-Golgi transport were independent of RicA and Rab2a, BspB-driven alterations in Golgi vesicular traffic also involved RicA and Rab2a, defining BspB and RicA's functional interplay at the Golgi interface. Altogether, these findings support a model where RicA modulation of Rab2a functions impairs replication but is compensated by BspB-mediated remodeling of Golgi apparatus-associated vesicular transport, revealing an epistatic interaction between these T4SS effectors. Bacterial pathogens with an intracellular lifestyle modulate many host cellular processes to promote their infectious cycle. They do so by delivering effector proteins into host cells via dedicated secretion systems that target specific host functions. While the roles of many individual effectors are known, how their modes of action are coordinated is rarely understood. Here, we show that the zoonotic bacterium delivers the BspB effector that mitigates the negative effect on bacterial replication that the RicA effector exerts via modulation of the host small GTPase Rab2. These findings provide an example of functional integration between bacterial effectors that promotes proliferation of pathogens.
胞内细菌病原体通过专门的分泌系统将效应分子输送到细胞内,从而在其感染周期中重塑细胞功能。虽然许多单个效应物的功能已被了解,但它们如何相互作用以促进发病机制却很少被理解。人畜共患病细菌 是布鲁氏菌病的病原体,通过其 VirB 型 IV 型分泌系统(T4SS)输送效应蛋白,介导内质网(ER)衍生的复制包含空泡(rBCV)的生物发生。在这里,我们表明 T4SS 效应物 BspB 和 RicA 在 复制中表现出上位性相互作用。由于缺失 而导致的 rBCV 生物发生和 复制缺陷依赖于宿主 GTPase Rab2a,并且缺失 可抑制这些表型缺陷,表明 Rab2 结合效应物 RicA 在这些表型缺陷中起作用。当缺失 和 时,rBCV 生物发生和细胞内复制对 Rab2a 的需求被消除,表明这些效应物的功能相互作用在 细胞内周期中涉及 Rab2 依赖性运输。RicA 的表达会损害宿主的分泌并导致高尔基体碎片化。虽然 BspB 介导的 ER 到高尔基体运输的改变与 RicA 和 Rab2a 无关,但 BspB 驱动的高尔基体囊泡运输的改变也涉及 RicA 和 Rab2a,这定义了 BspB 和 RicA 在高尔基体界面上的功能相互作用。总之,这些发现支持了这样一种模型,即 RicA 对 Rab2a 功能的调节会损害 的复制,但会被 BspB 介导的高尔基体器相关囊泡运输的重塑所补偿,揭示了这些 T4SS 效应物之间的上位性相互作用。具有细胞内生活方式的细菌病原体通过专门的分泌系统将效应蛋白输送到宿主细胞中,从而调节许多宿主细胞过程以促进其感染周期。它们通过靶向特定的宿主功能来实现这一点。虽然许多单个效应物的作用已被了解,但它们的作用方式如何协调却很少被理解。在这里,我们表明,人畜共患病细菌 输送 BspB 效应物,减轻 RicA 效应物通过调节宿主小 GTPase Rab2 对细菌复制的负面影响。这些发现提供了一个细菌效应物之间促进病原体增殖的功能整合的例子。