Iron Metabolism: Regulation and Diseases Group, Josep Carreras Leukaemia Research Institute (IJC), Campus Can Ruti, Badalona, 08916 Barcelona, Spain.
EuroBloodNet Referral Center for Iron Disorders and Gruppo Interdisciplinare Malattie del Ferro, Internal Medicine Unit, Azienda Ospedaliera Universitaria Integrata di Verona, 37134 Verona, Italy.
Int J Mol Sci. 2020 Mar 30;21(7):2374. doi: 10.3390/ijms21072374.
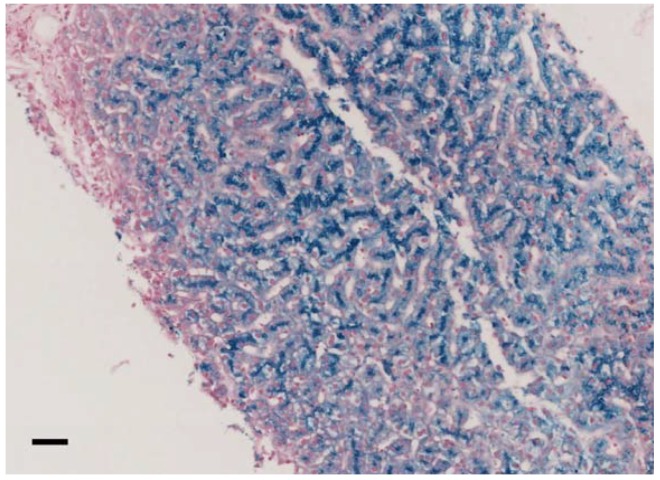
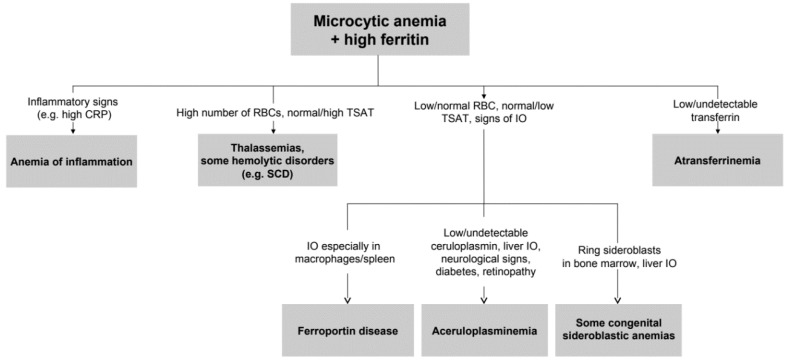
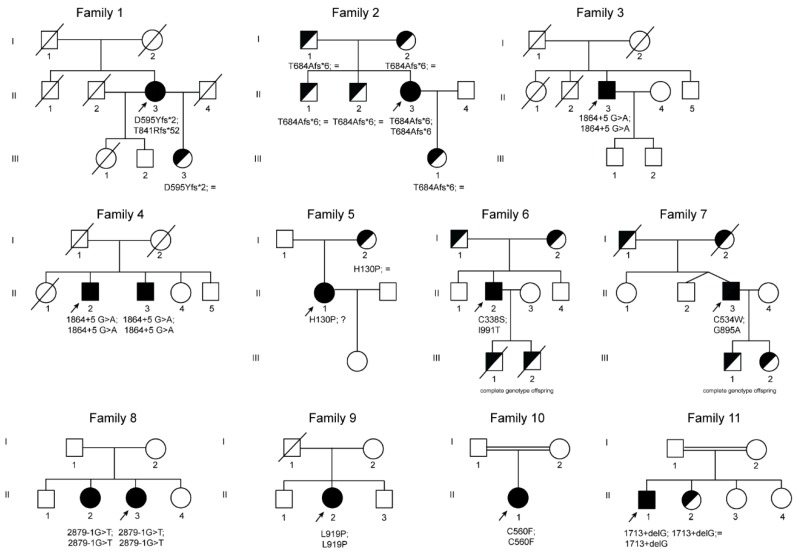
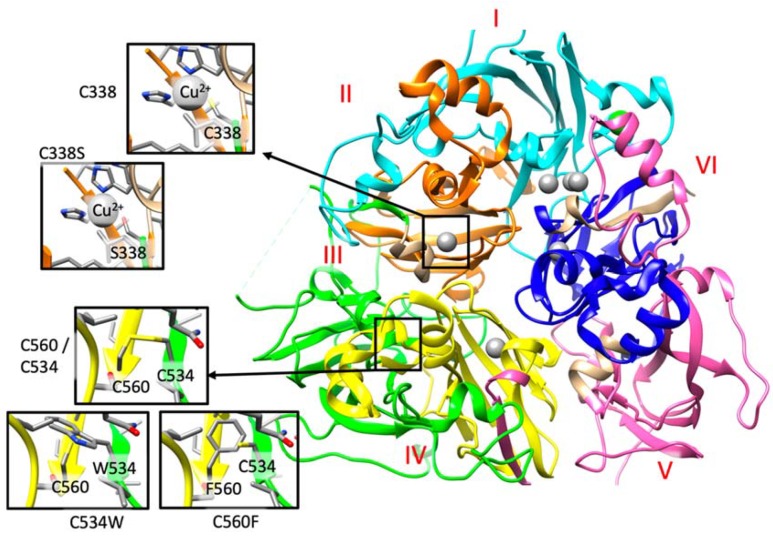
Aceruloplasminemia is a rare autosomal recessive genetic disease characterized by mild microcytic anemia, diabetes, retinopathy, liver disease, and progressive neurological symptoms due to iron accumulation in pancreas, retina, liver, and brain. The disease is caused by mutations in the Ceruloplasmin () gene that produce a strong reduction or absence of ceruloplasmin ferroxidase activity, leading to an impairment of iron metabolism. Most patients described so far are from Japan. Prompt diagnosis and therapy are crucial to prevent neurological complications since, once established, they are usually irreversible. Here, we describe the largest series of non-Japanese patients with aceruloplasminemia published so far, including 13 individuals from 11 families carrying 13 mutations in the gene (7 missense, 3 frameshifts, and 3 splicing mutations), 10 of which are novel. All missense mutations were studied by computational modeling. Clinical manifestations were heterogeneous, but anemia, often but not necessarily microcytic, was frequently the earliest one. This study confirms the clinical and genetic heterogeneity of aceruloplasminemia, a disease expected to be increasingly diagnosed in the Next-Generation Sequencing (NGS) era. Unexplained anemia with low transferrin saturation and high ferritin levels without inflammation should prompt the suspicion of aceruloplasminemia, which can be easily confirmed by low serum ceruloplasmin levels. Collaborative joint efforts are needed to better understand the pathophysiology of this potentially disabling disease.
亚铁氧化酶蛋白血症是一种罕见的常染色体隐性遗传性疾病,其特征为轻度小细胞性贫血、糖尿病、视网膜病变、肝病以及由于铁在胰腺、视网膜、肝脏和大脑中蓄积导致的进行性神经症状。该疾病由载脂蛋白(Ceruloplasmin (AP))基因的突变引起,这些突变导致亚铁氧化酶活性明显降低或缺失,从而导致铁代谢紊乱。迄今为止,大多数描述的患者都来自日本。及时诊断和治疗对于预防神经并发症至关重要,因为一旦发生,这些并发症通常是不可逆的。在这里,我们描述了迄今为止发表的最大系列的非日本亚铁氧化酶蛋白血症患者,包括来自 11 个家庭的 13 名个体,这些个体携带载脂蛋白(AP)基因中的 13 种突变(7 种错义突变、3 种移码突变和 3 种剪接突变),其中 10 种是新的。所有错义突变均通过计算建模进行了研究。临床表现具有异质性,但贫血(通常但并非一定为小细胞性贫血)常常是最早出现的症状之一。本研究证实了亚铁氧化酶蛋白血症的临床和遗传异质性,在下一代测序(Next-Generation Sequencing (NGS))时代,预计该疾病的诊断会越来越多。如果出现原因不明的贫血、低转铁蛋白饱和度和高铁蛋白水平而无炎症,应怀疑为亚铁氧化酶蛋白血症,通过检测血清中低水平的亚铁氧化酶蛋白可轻易确诊。需要协作努力以更好地了解这种潜在致残疾病的病理生理学。