Taylor James O, Neri Gaia, Banerji Liam, Cowan Alexander J, Hartl František
Department of Chemistry, University of Reading, Reading RG6 6AD, United Kingdom.
Department of Chemistry, Stephenson Institute for Renewable Energy, University of Liverpool, Liverpool L69 7ZF, United Kingdom.
Inorg Chem. 2020 Apr 20;59(8):5564-5578. doi: 10.1021/acs.inorgchem.0c00263. Epub 2020 Apr 2.
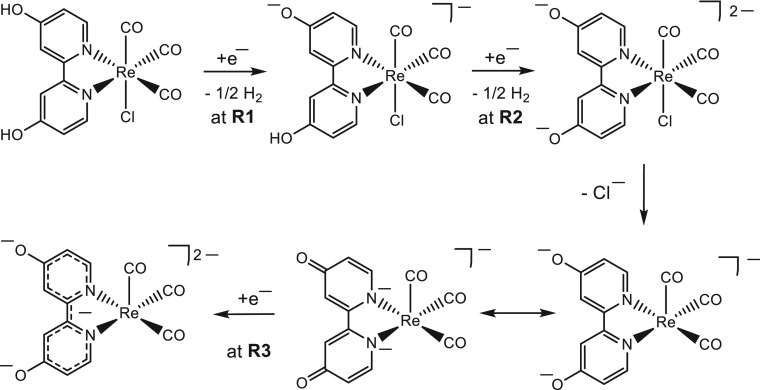
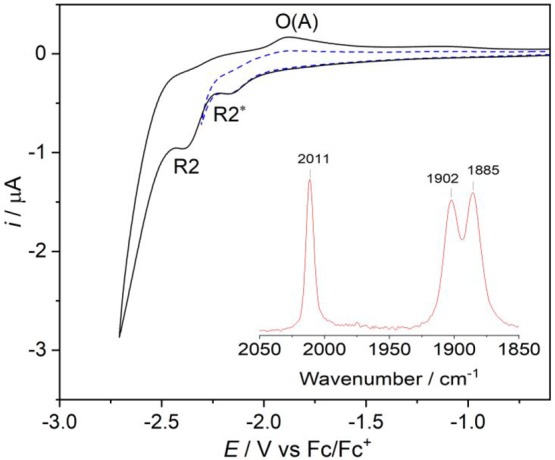
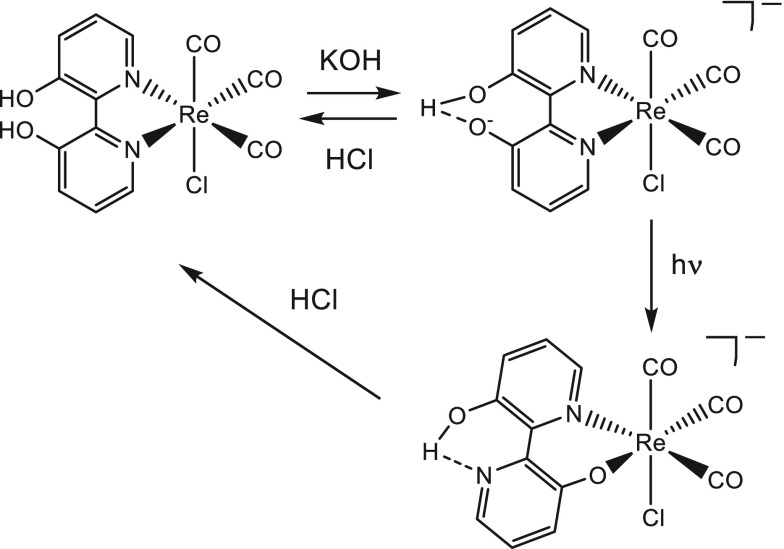
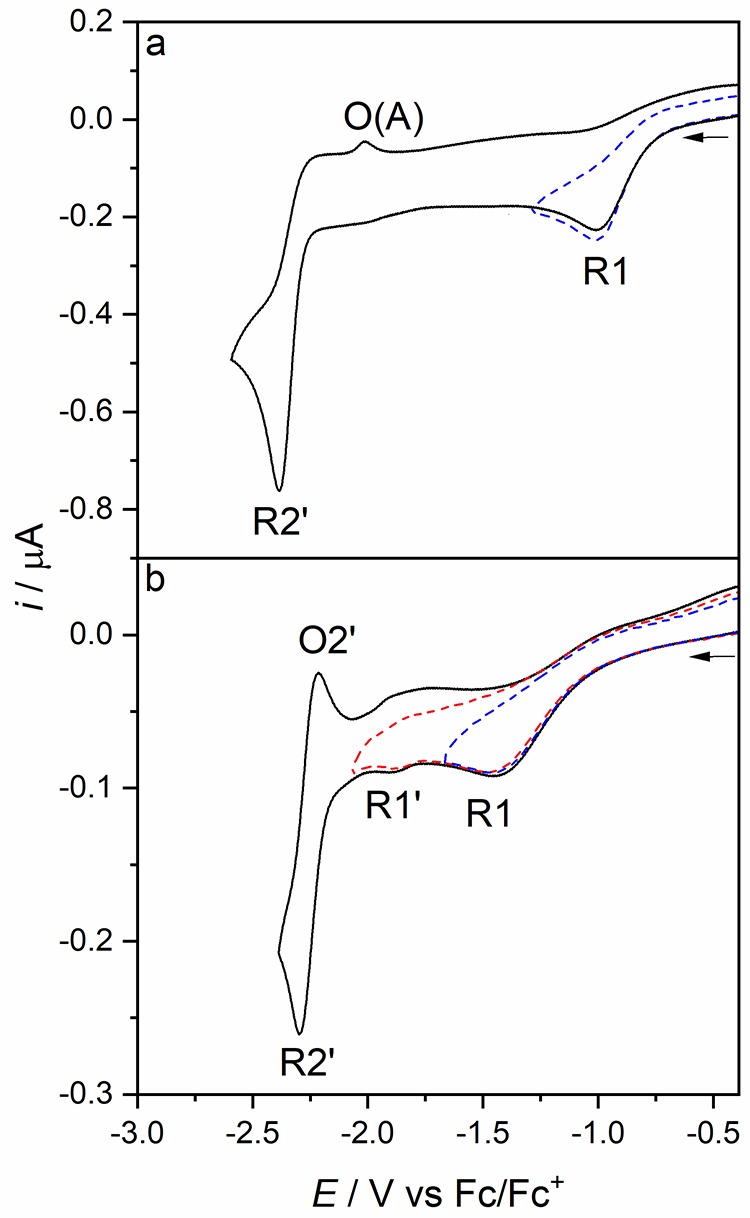
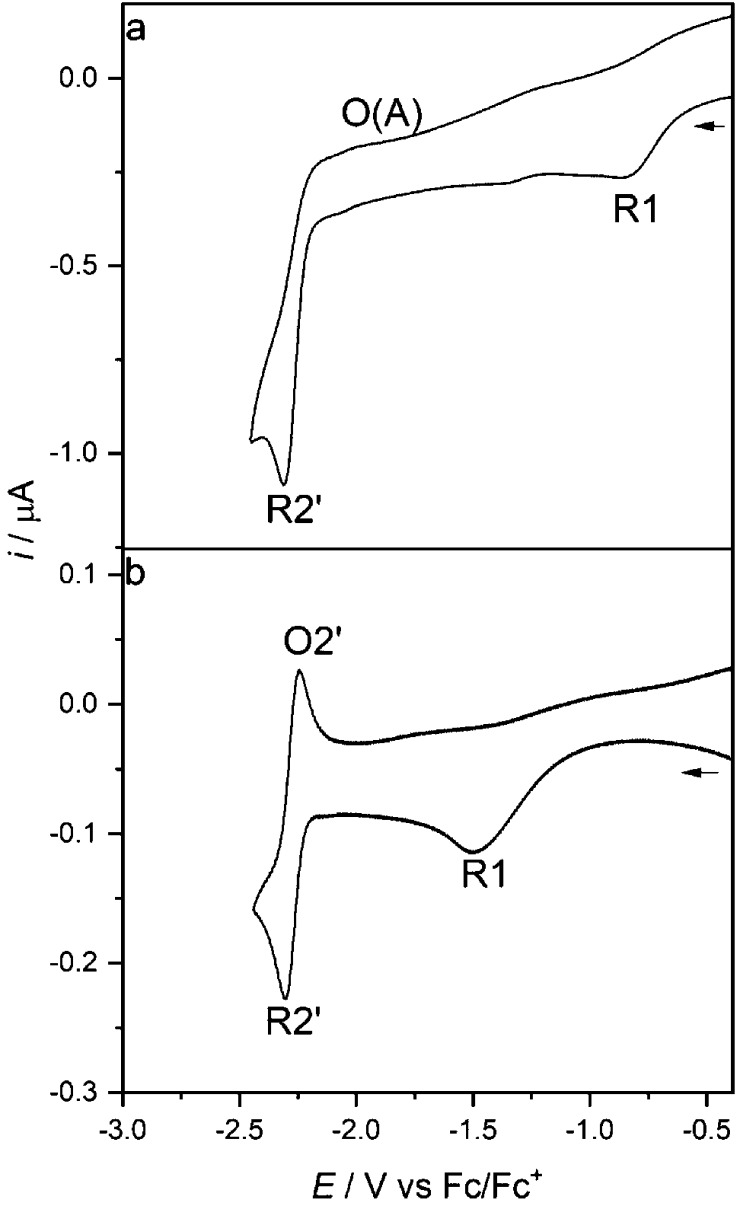
Herein, we present the cathodic paths of the Group-7 metal complex [Re(3,3'-DHBPY)(CO)Cl] (3,3'-DHBPY = 3,3'-dihydroxy-2,2'-bipyridine) producing a moderately active catalyst of electrochemical reduction of CO to CO. The combined techniques of cyclic voltammetry and IR/UV-vis spectroelectrochemistry have revealed significant differences in the chemistry of the electrochemically reduced parent complex compared to the previously published Re/4,4'-DHBPY congener. The initial irreversible cathodic step in weakly coordinating THF is shifted toward much less negative electrode potentials, reflecting facile reductive deprotonation of one hydroxyl group and strong intramolecular hydrogen bonding, O-H···O. The latter process occurs spontaneously in basic dimethylformamide where Re/4,4'-DHBPY remains stable. The subsequent reduction of singly deprotonated [Re(3,3'-DHBPY-H)(CO)Cl] under ambient conditions occurs at a cathodic potential close to that of the Re/4,4'-DHBPY-H derivative. However, for the stabilized 3,3'-DHBPY-H ligand, the latter process at the second cathodic wave is more complex and involves an overall transfer of three electrons. Rapid potential step electrolysis induces 1e-reductive cleavage of the second O-H bond, triggering dissociation of the Cl ligand from [Re(3,3'-DHBPY-2H)(CO)Cl]. The ultimate product of the second cathodic step in THF was identified as 5-coordinate [Re(3,3'-DHBPY-2H)(CO)], the equivalent of classical 2e-reduced [Re(BPY)(CO)]. Each reductive deprotonation of the DHBPY ligand results in a redshift of the IR ν(CO) absorption of the tricarbonyl complexes by ca. 10 cm, facilitating the product assignment based on comparison with the literature data for corresponding Re/BPY complexes. The Cl dissociation from [Re(3,3'-DHBPY-2H)(CO)Cl] was proven in strongly coordinating butyronitrile. The latter dianion is stable at 223 K, converting at 258 K to 6-coordinate [Re(3,3'-DHBPY-2H)(CO)(PrCN)]. Useful reference data were obtained with substituted parent [Re(3,3'-DHBPY)(CO)(PrCN)] that also smoothly deprotonates by the initial reduction to [Re(3,3'-DHBPY-H)(CO)(PrCN)]. The latter complex ultimately converts at the second cathodic wave to [Re(3,3'-DHBPY-2H)(CO)(PrCN)] via a counterintuitive ETC step generating the 1e radical of the parent complex, viz., [Re(3,3'-DHBPY)(CO)(PrCN)]. The same alternative reduction path is also followed by [Re(3,3'-DHBPY-H)(CO)Cl] at the onset of the second cathodic wave, where the ETC step results in the intermediate [Re(3,3'-DHBPY)(CO)Cl] further reducible to [Re(3,3'-DHBPY-2H)(CO)] as the CO catalyst.
在此,我们展示了第7族金属配合物[Re(3,3'-DHBPY)(CO)Cl](3,3'-DHBPY = 3,3'-二羟基-2,2'-联吡啶)的阴极路径,该配合物可产生一种将CO电化学还原为CO的中等活性催化剂。循环伏安法和红外/紫外-可见光谱电化学的联合技术揭示了与先前发表的Re/4,4'-DHBPY同系物相比,电化学还原的母体配合物在化学性质上存在显著差异。在弱配位的四氢呋喃中,最初的不可逆阴极步骤向负得多的电极电位偏移,这反映了一个羟基的容易的还原去质子化以及强的分子内氢键O-H···O。后一过程在碱性二甲基甲酰胺中自发发生,而Re/4,4'-DHBPY在其中保持稳定。在环境条件下,单去质子化的[Re(3,3'-DHBPY-H)(CO)Cl]的后续还原发生在接近Re/4,4'-DHBPY-H衍生物的阴极电位处。然而,对于稳定化的3,3'-DHBPY-H配体,在第二个阴极波处的后一过程更为复杂,涉及三个电子的整体转移。快速电位阶跃电解诱导第二个O-H键的1e还原裂解,引发Cl配体从[Re(3,3'-DHBPY-2H)(CO)Cl]上解离。在四氢呋喃中第二个阴极步骤的最终产物被鉴定为五配位的[Re(3,3'-DHBPY-2H)(CO)],相当于经典的2e还原的[Re(BPY)(CO)]。DHBPY配体的每次还原去质子化都会使三羰基配合物的红外ν(CO)吸收发生约10 cm的红移,便于根据与相应Re/BPY配合物的文献数据比较来进行产物归属。在强配位的丁腈中证实了Cl从[Re(3,3'-DHBPY-2H)(CO)Cl]上解离。后一种二价阴离子在223 K时稳定,在258 K时转化为六配位的[Re(3,3'-DHBPY-2H)(CO)(PrCN)]。用取代的母体[Re(3,3'-DHBPY)(CO)(PrCN)]获得了有用的参考数据,该母体也通过初始还原顺利去质子化为[Re(3,3'-DHBPY-H)(CO)(PrCN)]。后一种配合物最终在第二个阴极波处通过一个违反直觉的电子转移步骤转化为[Re(3,3'-DHBPY-2H)(CO)(PrCN)],该步骤产生母体配合物的1e自由基,即[Re(3,3'-DHBPY)(CO)(PrCN)]。在第二个阴极波开始时,[Re(3,3'-DHBPY-H)(CO)Cl]也遵循相同的替代还原路径,其中电子转移步骤产生中间体[Re(3,3'-DHBPY)(CO)Cl],该中间体可进一步还原为作为CO催化剂的[Re(3,3'-DHBPY-2H)(CO)]。