Department of Pharmaceutical Toxicology, Faculty of Pharmacy, Istanbul University, Istanbul, Turkey; Department of Biochemistry, School of Medicine / Genetic and Metabolic Diseases Research and Investigation Center, Marmara University, Istanbul, Turkey.
Department of Biochemistry, School of Medicine / Genetic and Metabolic Diseases Research and Investigation Center, Marmara University, Istanbul, Turkey.
Redox Biol. 2020 May;32:101502. doi: 10.1016/j.redox.2020.101502. Epub 2020 Mar 21.
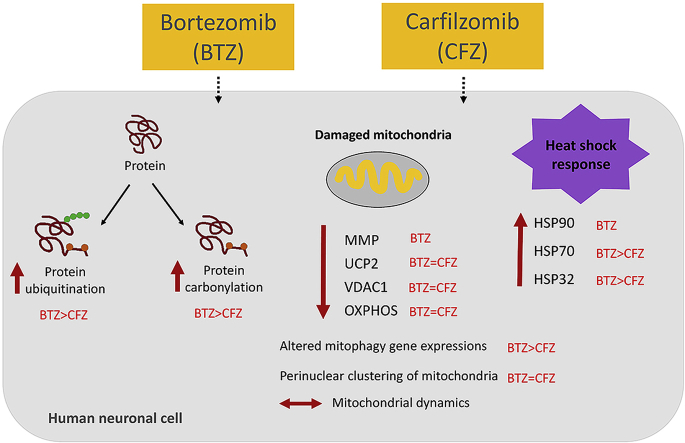
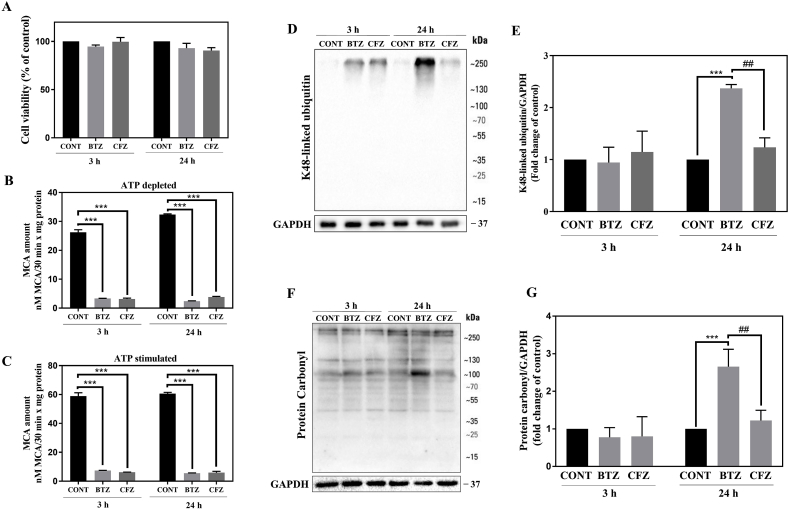
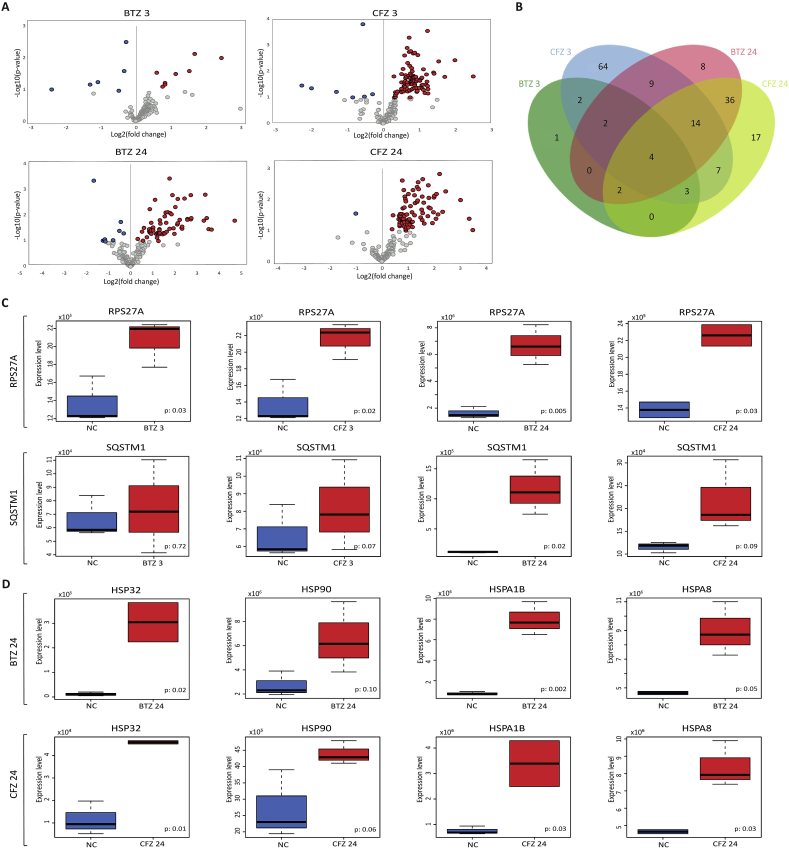
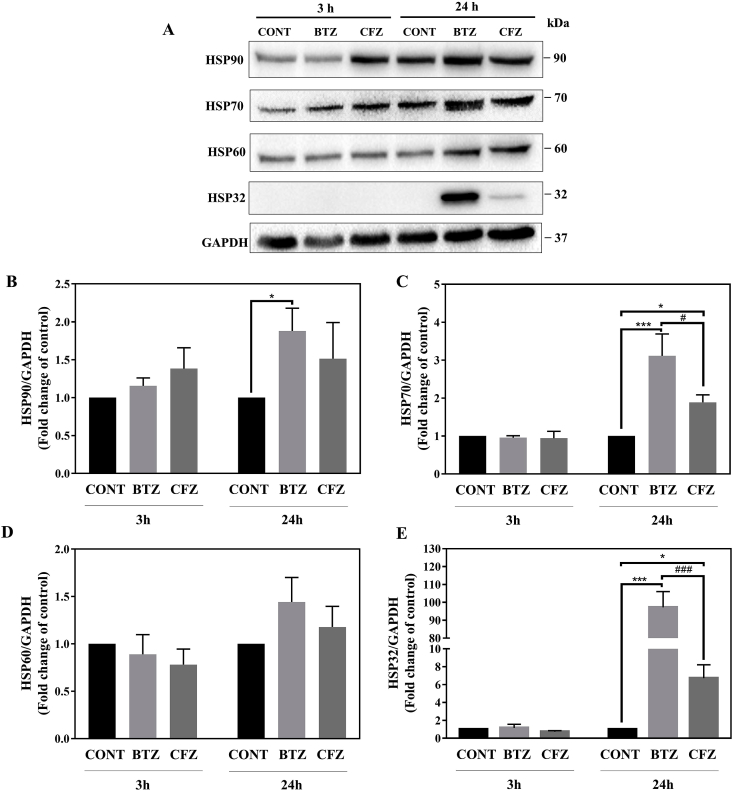
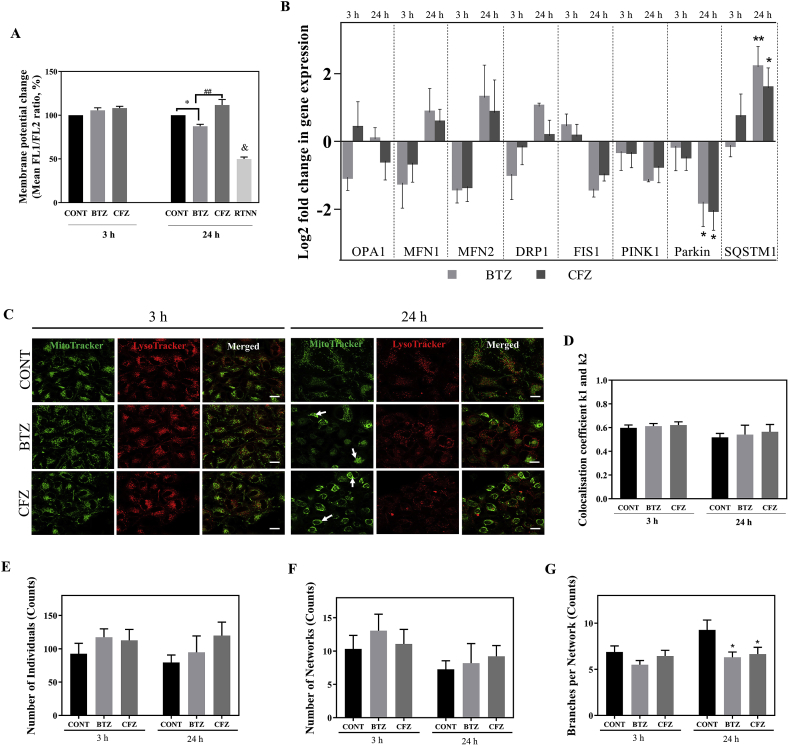
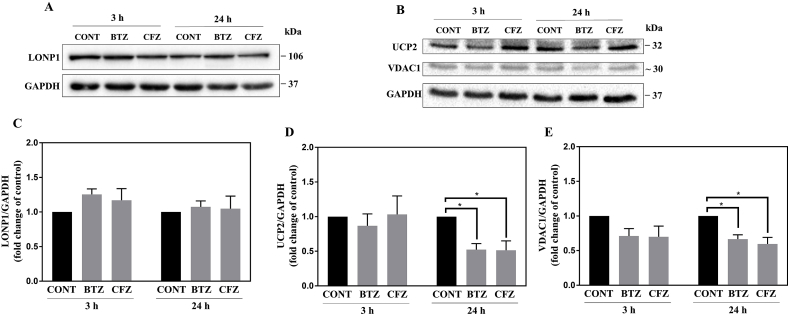
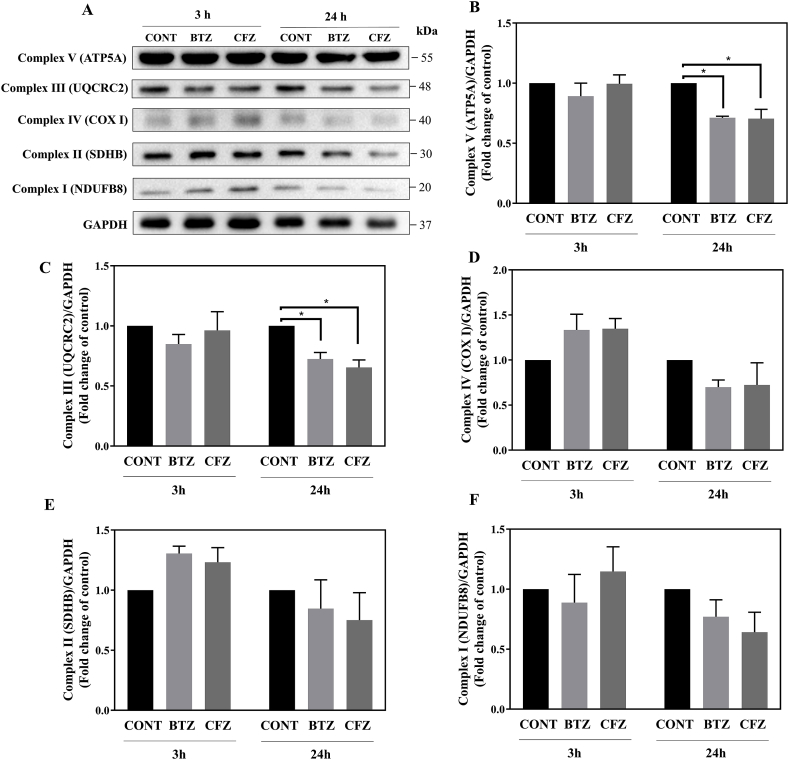
Proteasome inhibitors have great success for their therapeutic potential against hematologic malignancies. First generation proteasome inhibitor bortezomib induced peripheral neuropathy is considered as a limiting factor in chemotherapy and its second-generation counterpart carfilzomib is associated with lower rates of neurotoxicity. The mitochondrial toxicity (mitotoxicity) hypothesis arises from studies with animal models of bortezomib induced peripheral neuropathy. However, molecular mechanisms are not fully elucidated and the role of mitotoxicity in bortezomib and carfilzomib induced neurotoxicity has not been investigated comparatively. Herein, we characterized the neurotoxic effects of bortezomib and carfilzomib at the molecular level in human neuronal cells using LC-MS/MS analysis, flow cytometry, RT-qPCR, confocal microscopy and western blotting. We showed that bortezomib and carfilzomib affected the human neuronal proteome differently, and bortezomib caused higher proteotoxic stress via protein oxidation, protein K48-ubiquitination, heat shock protein expression upregulation and reduction of mitochondria membrane potential. Bortezomib and carfilzomib did not affect the gene expression levels related to mitochondrial dynamics (optic atrophy 1; OPA1, mitofusin 1; MFN1, mitofusin 2; MFN2, fission 1; FIS1, dynamin-related protein 1; DRP1) and overall mitophagy rate whereas, PINK1/Parkin mediated mitophagy gene expressions were altered with both drugs. Bortezomib and carfilzomib caused downregulation of the contents of mitochondrial oxidative phosphorylation complexes, voltage-dependent anion channel 1 (VDAC1) and uncoupling protein 2 (UCP2) similarly. Our findings suggest that, both drugs induce mitotoxicity besides proteotoxic stress in human neuronal cells and the higher incidence of neurotoxicity with bortezomib than carfilzomib is not directly related to mitochondrial pathways.
蛋白酶体抑制剂在治疗血液系统恶性肿瘤方面具有巨大的治疗潜力。第一代蛋白酶体抑制剂硼替佐米引起的周围神经病被认为是化疗的一个限制因素,而其第二代同类药物卡非佐米则与较低的神经毒性发生率相关。线粒体毒性(mitotoxicity)假说源于硼替佐米诱导的周围神经病动物模型的研究。然而,分子机制尚未完全阐明,并且线粒体毒性在硼替佐米和卡非佐米诱导的神经毒性中的作用尚未得到比较研究。在此,我们使用 LC-MS/MS 分析、流式细胞术、RT-qPCR、共聚焦显微镜和 Western blot 技术在人神经元细胞中从分子水平表征了硼替佐米和卡非佐米的神经毒性作用。我们表明,硼替佐米和卡非佐米对人神经元蛋白质组的影响不同,并且硼替佐米通过蛋白质氧化、蛋白质 K48-泛素化、热休克蛋白表达上调和线粒体膜电位降低引起更高的蛋白毒性应激。硼替佐米和卡非佐米不影响与线粒体动力学相关的基因表达水平(视神经萎缩 1;OPA1、线粒体融合蛋白 1;MFN1、线粒体融合蛋白 2;MFN2、分裂 1;FIS1、动力相关蛋白 1;DRP1)和整体线粒体自噬率,而两种药物都改变了 PINK1/Parkin 介导的线粒体自噬基因表达。硼替佐米和卡非佐米引起线粒体氧化磷酸化复合物、电压依赖性阴离子通道 1(VDAC1)和解偶联蛋白 2(UCP2)含量的下调相似。我们的研究结果表明,这两种药物除了在人神经元细胞中引起蛋白毒性应激外,还引起线粒体毒性,而硼替佐米比卡非佐米引起更高的神经毒性发生率与线粒体途径没有直接关系。