Department of Pediatrics, Cincinnati Children's Hospital Medical Center, University of Cincinnati, 3333 Burnet Avenue, MLC 7037, Cincinnati, OH, 45229-3026, USA.
Department of Electrical Engineering and Computer Science, University of Cincinnati, Cincinnati, OH, 45221, USA.
BMC Bioinformatics. 2020 Apr 3;21(1):131. doi: 10.1186/s12859-020-3462-5.
Admixed populations arise when two or more previously isolated populations interbreed. A powerful approach to addressing the genetic complexity in admixed populations is to infer ancestry. Ancestry inference including the proportion of an individual's genome coming from each population and its ancestral origin along the chromosome of an admixed population requires the use of ancestry informative markers (AIMs) from reference ancestral populations. AIMs exhibit substantial differences in allele frequency between ancestral populations. Given the huge amount of human genetic variation data available from diverse populations, a computationally feasible and cost-effective approach is becoming increasingly important to extract or filter AIMs with the maximum information content for ancestry inference, admixture mapping, forensic applications, and detecting genomic regions that have been under recent selection.
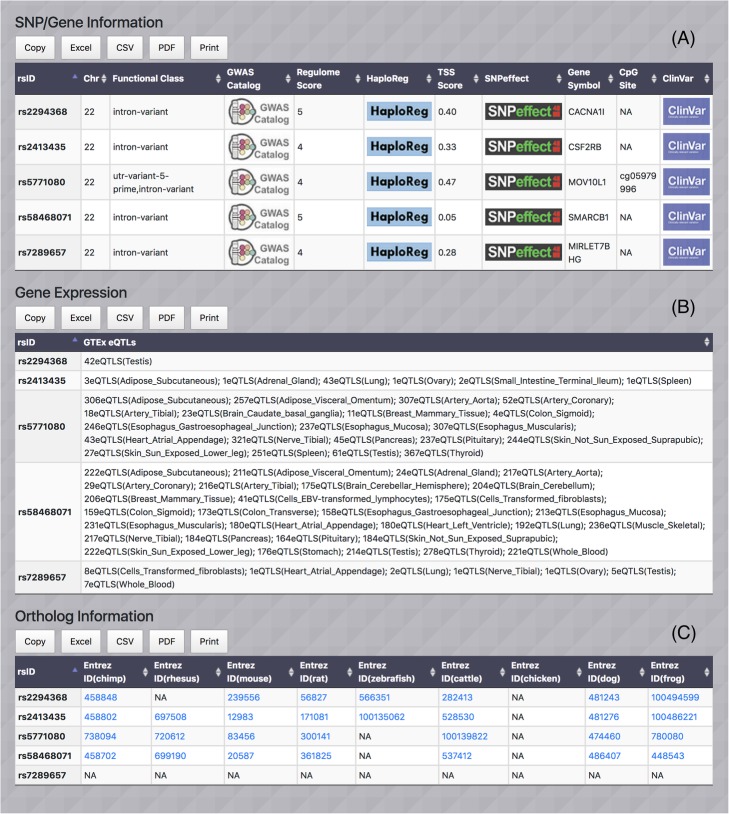
To address this gap, we present MI-MAAP, an easy-to-use web-based bioinformatics tool designed to prioritize informative markers for multi-ancestry admixed populations by utilizing feature selection methods and multiple genomics resources including 1000 Genomes Project and Human Genome Diversity Project. Specifically, this tool implements a novel allele frequency-based feature selection algorithm, Lancaster Estimator of Independence (LEI), as well as other genotype-based methods such as Principal Component Analysis (PCA), Support Vector Machine (SVM), and Random Forest (RF). We demonstrated that MI-MAAP is a useful tool in prioritizing informative markers and accurately classifying ancestral populations. LEI is an efficient feature selection strategy to retrieve ancestry informative variants with different allele frequency/selection pressure among (or between) ancestries without requiring computationally expensive individual-level genotype data.
MI-MAAP has a user-friendly interface which provides researchers an easy and fast way to filter and identify AIMs. MI-MAAP can be accessed at https://research.cchmc.org/mershalab/MI-MAAP/login/.
当两个或更多以前隔离的群体杂交时,就会出现混合群体。解决混合群体遗传复杂性的一种有力方法是推断其祖先。推断祖先包括个体基因组中来自每个群体的比例及其在混合群体中的染色体祖先起源,这需要使用来自参考祖先群体的祖先信息标记 (AIMs)。AIMs 在等位基因频率方面在祖先群体之间存在显著差异。鉴于来自不同群体的大量人类遗传变异数据可用,提取或过滤具有最大祖先推断、混合映射、法医应用和检测最近受到选择的基因组区域信息量的 AIMs 的计算上可行且具有成本效益的方法变得越来越重要。
为了解决这一差距,我们提出了 MI-MAAP,这是一种易于使用的基于网络的生物信息学工具,旨在通过利用特征选择方法和多种基因组学资源(包括 1000 基因组计划和人类基因组多样性计划),优先为多祖先混合群体选择信息标记。具体来说,该工具实现了一种新颖的基于等位基因频率的特征选择算法,即兰开斯特独立性估计器 (LEI),以及其他基于基因型的方法,如主成分分析 (PCA)、支持向量机 (SVM) 和随机森林 (RF)。我们证明了 MI-MAAP 是一种有用的工具,可以优先选择信息标记并准确分类祖先群体。LEI 是一种有效的特征选择策略,可在不要求计算昂贵的个体水平基因型数据的情况下,从(或在)祖先之间检索具有不同等位基因频率/选择压力的祖先信息变体。
MI-MAAP 具有用户友好的界面,为研究人员提供了一种简单快捷的方法来筛选和识别 AIMs。MI-MAAP 可在 https://research.cchmc.org/mershalab/MI-MAAP/login/ 访问。