Wang Yuxin, Ouyang Meishuo, Gao Xibao, Wang Shuai, Fu Chunyang, Zeng Jiayi, He Xiaodong
Department of Physical and Chemical Inspection, School of Public Health, Shandong University, Jinan, Shandong 250012, People's Republic of China.
Department of Public Health, Graduate School of Medicine, Osaka University, Suita, Osaka 565-0871, Japan.
Diabetes Metab Syndr Obes. 2020 Mar 19;13:835-850. doi: 10.2147/DMSO.S240728. eCollection 2020.
The purpose of this study was to explore the difference and association between intestinal microbiota and plasma metabolomics between type 2 diabetes mellitus (T2DM) and normal group and to identify potential microbiota biomarkers that contribute the most to the difference in metabolites.
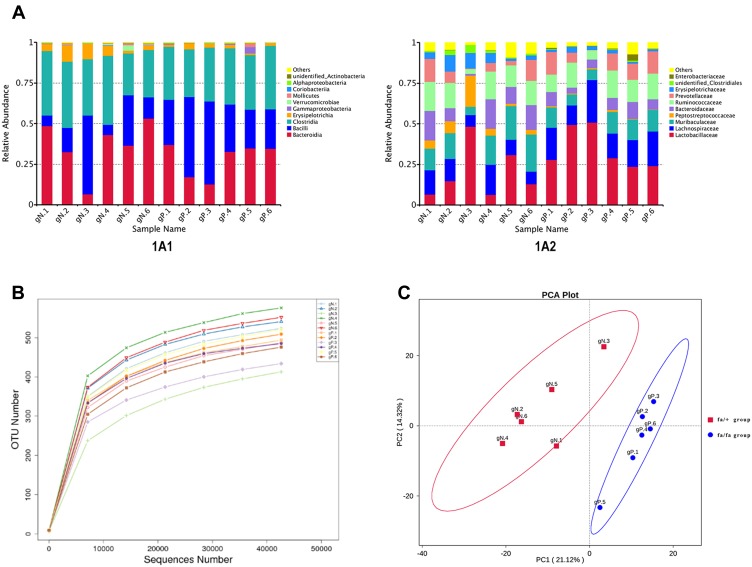
Six male ZDF model (fa/fa) rats were fed by a Purina #5008 Lab Diet (crude protein 23.5%, crude fat 6.5%) for 3 weeks and their age-matched 6 ZDF control (fa/+) rats were fed by normal rodent diet. Their stool and blood samples were collected at 12 weeks. To analyze the microbial populations in these samples, we used a 16S rRNA gene sequencing approach. Liquid chromatography-mass spectrometry (LC-MS) followed by multivariate statistical analysis was applied to the plasma metabolites profiling. Correlation analysis of them was calculated by Pearson statistical method.
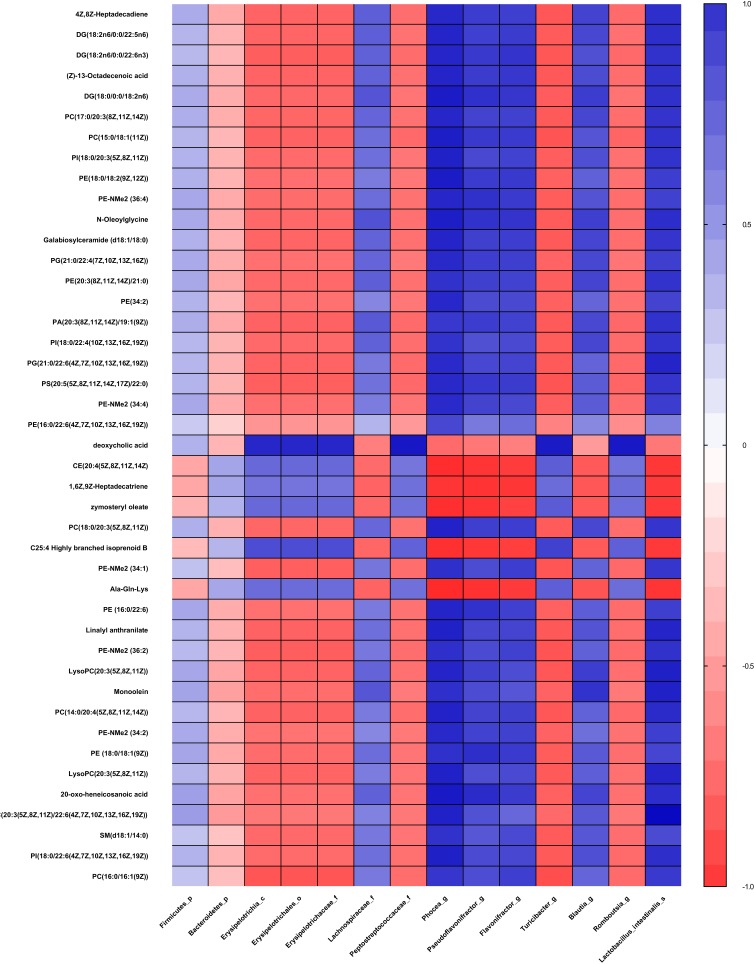
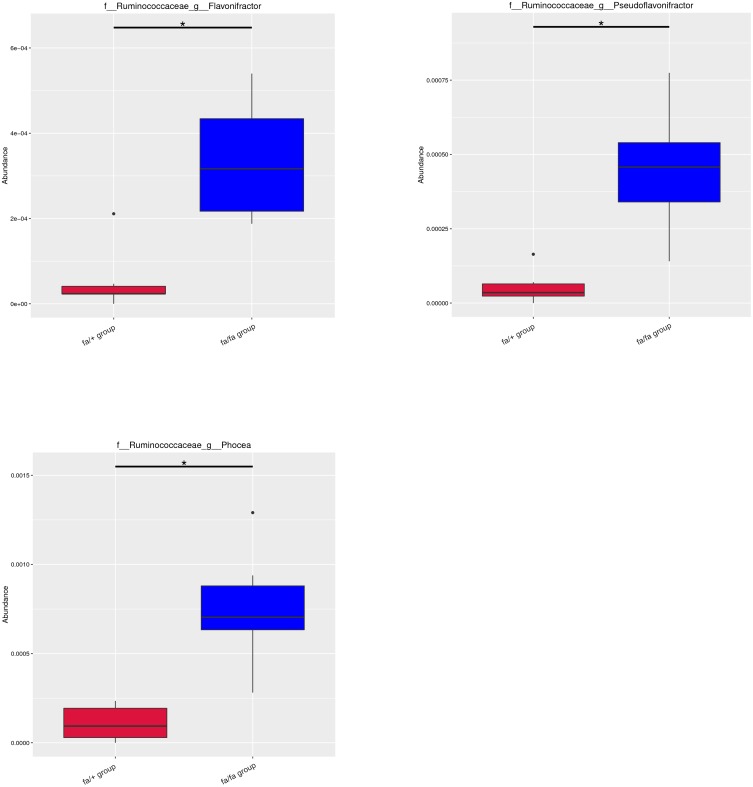
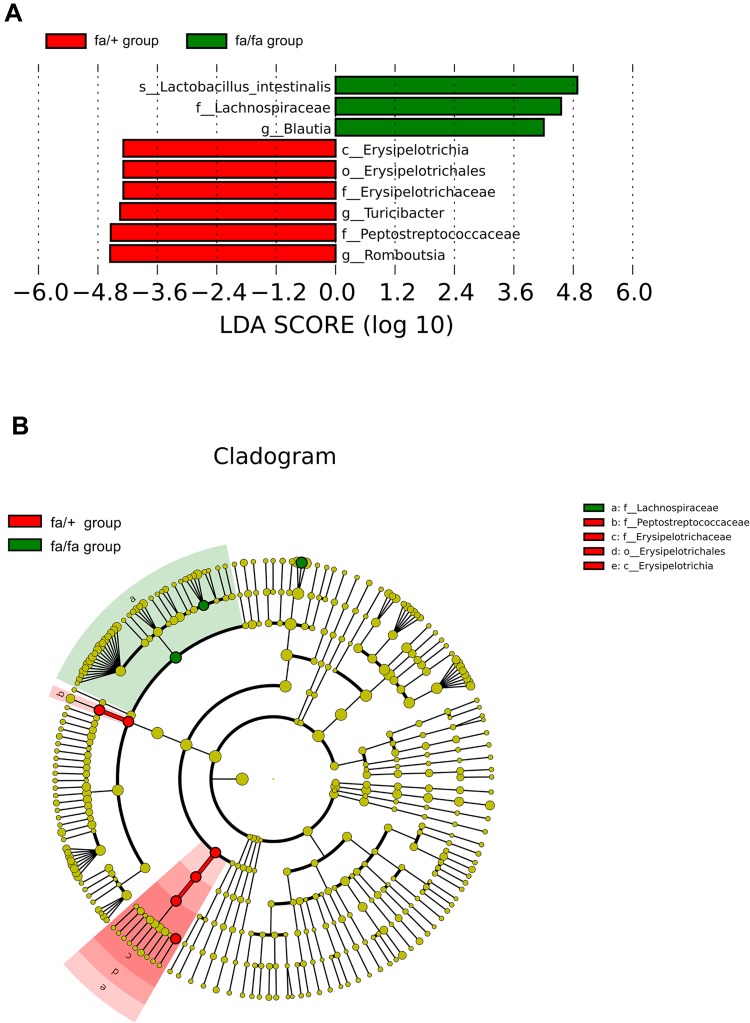
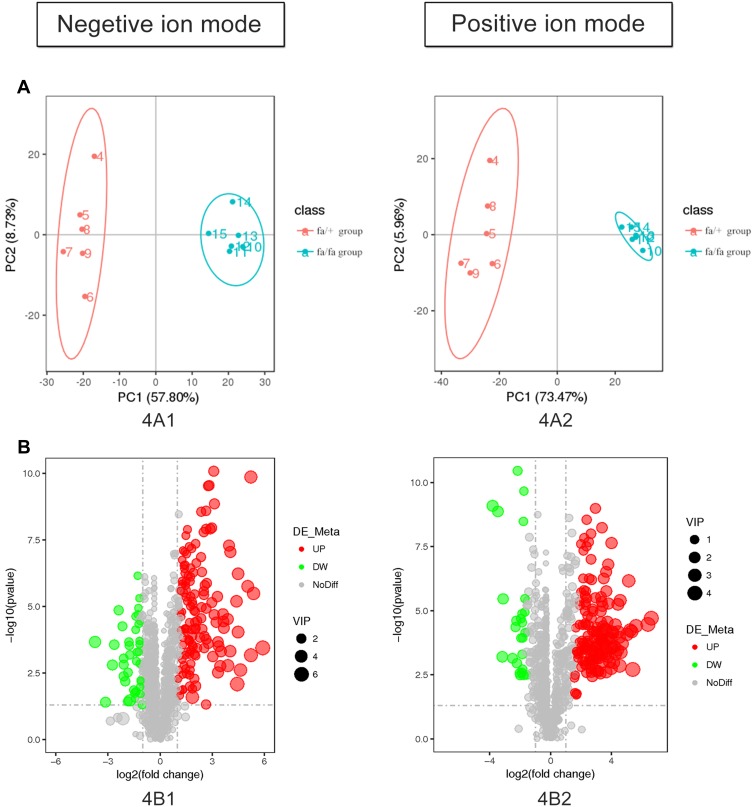
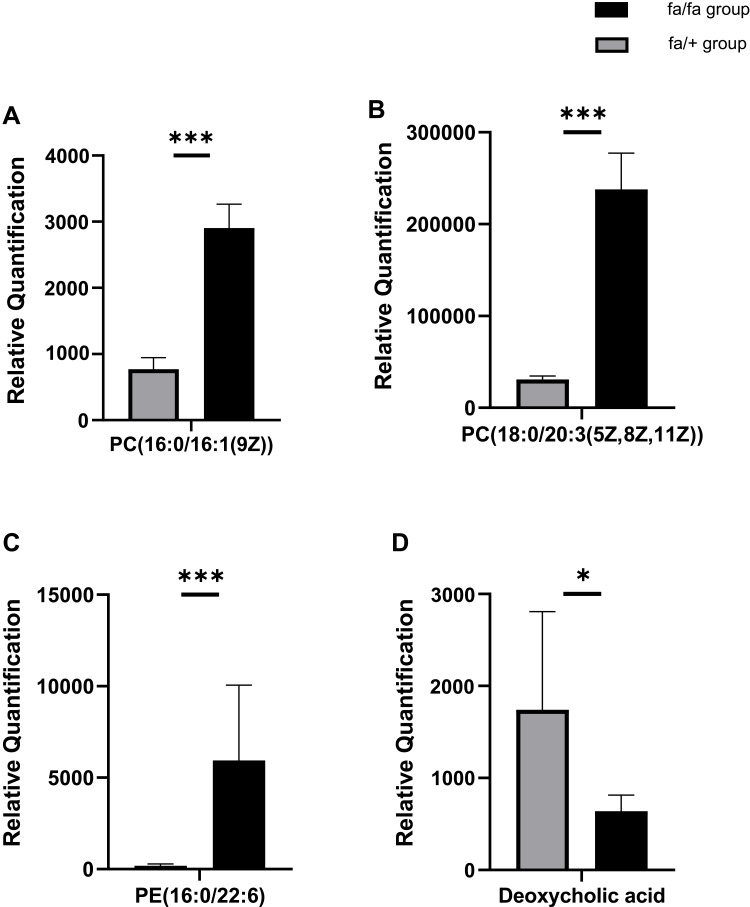
Twelve potential biomarkers of intestinal microbial flora and 357 differential metabolites were found in ZDF fa/fa rats, among which there are three flora that contributed the most to the perturbation of metabolites, including genus and species .
Our study demonstrates the alterations of the abundance and diversity of the intestinal microbiota and the perturbation of metabolites in ZDF rats (fa/fa). We found three potential biomarkers of intestinal microbiota that may lead to perturbation in plasma metabolites. This may prompt new pathogenesis of obesity-related T2DM, but we also need to study further about the causal relationship between intestinal microbe and T2DM, so as to find the target of T2DM treatment or preventive measures.
本研究旨在探讨2型糖尿病(T2DM)组与正常组之间肠道微生物群与血浆代谢组学的差异及关联,并确定对代谢物差异贡献最大的潜在微生物生物标志物。
6只雄性ZDF模型(fa/fa)大鼠用普瑞纳#5008实验饲料(粗蛋白23.5%,粗脂肪6.5%)喂养3周,6只年龄匹配的ZDF对照(fa/+)大鼠用正常啮齿动物饲料喂养。在12周时采集它们的粪便和血液样本。为了分析这些样本中的微生物种群,我们采用了16S rRNA基因测序方法。采用液相色谱-质谱联用(LC-MS)并结合多变量统计分析对血浆代谢物进行分析。通过Pearson统计方法计算它们之间的相关性分析。
在ZDF fa/fa大鼠中发现了12种潜在的肠道微生物群生物标志物和357种差异代谢物,其中有三种菌群对代谢物的扰动贡献最大,包括属和种。
我们的研究证明了ZDF大鼠(fa/fa)肠道微生物群丰度和多样性的改变以及代谢物的扰动。我们发现了三种可能导致血浆代谢物扰动的肠道微生物群潜在生物标志物。这可能提示肥胖相关T2DM的新发病机制,但我们还需要进一步研究肠道微生物与T2DM之间的因果关系,以便找到T2DM治疗靶点或预防措施。