Intelligent Systems Laboratory, University of Bristol, Bristol BS8 1UB, UK.
MRC Integrative Epidemiology Unit (IEU), University of Bristol, Bristol BS8 2BN, UK.
Bioinformatics. 2020 Jun 1;36(12):3637-3644. doi: 10.1093/bioinformatics/btaa242.
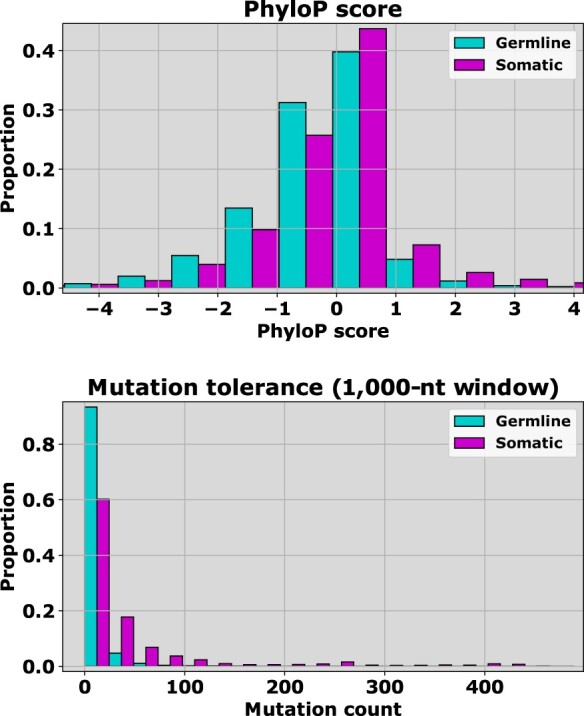
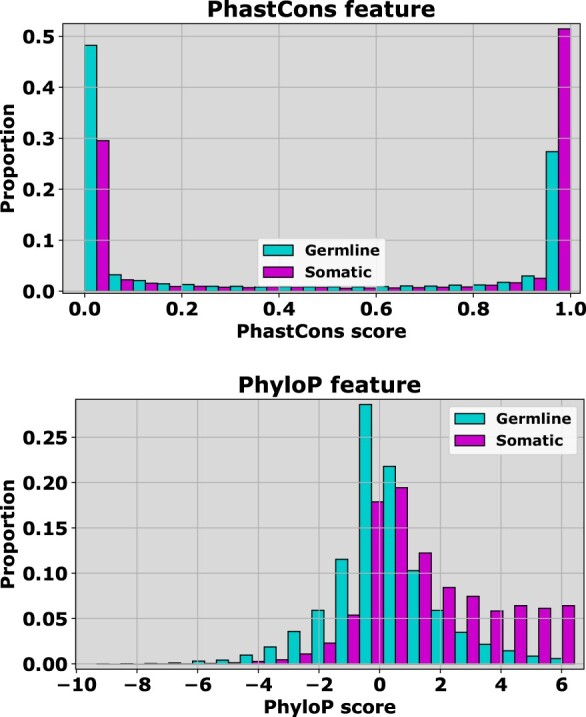
Next-generation sequencing technologies have accelerated the discovery of single nucleotide variants in the human genome, stimulating the development of predictors for classifying which of these variants are likely functional in disease, and which neutral. Recently, we proposed CScape, a method for discriminating between cancer driver mutations and presumed benign variants. For the neutral class, this method relied on benign germline variants found in the 1000 Genomes Project database. Discrimination could, therefore, be influenced by the distinction of germline versus somatic, rather than neutral versus disease driver. This motivates this article in which we consider predictive discrimination between recurrent and rare somatic single point mutations based solely on using cancer data, and the distinction between these two somatic classes and germline single point mutations.
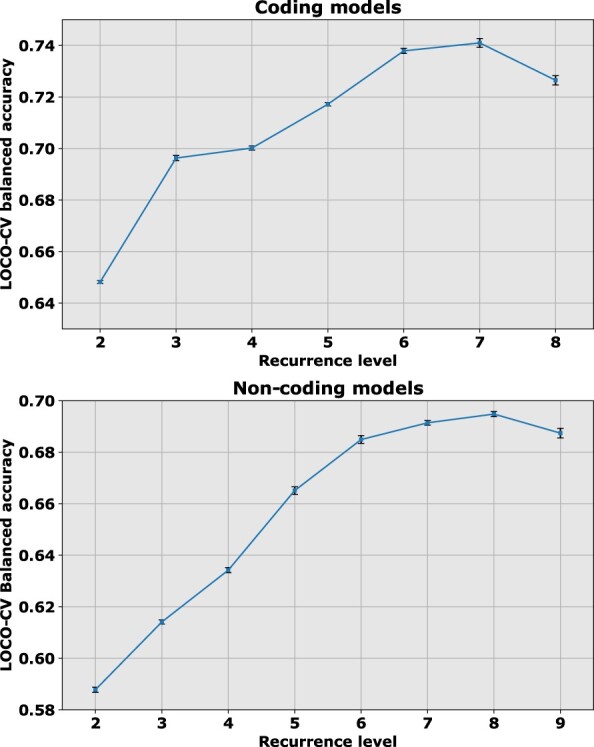
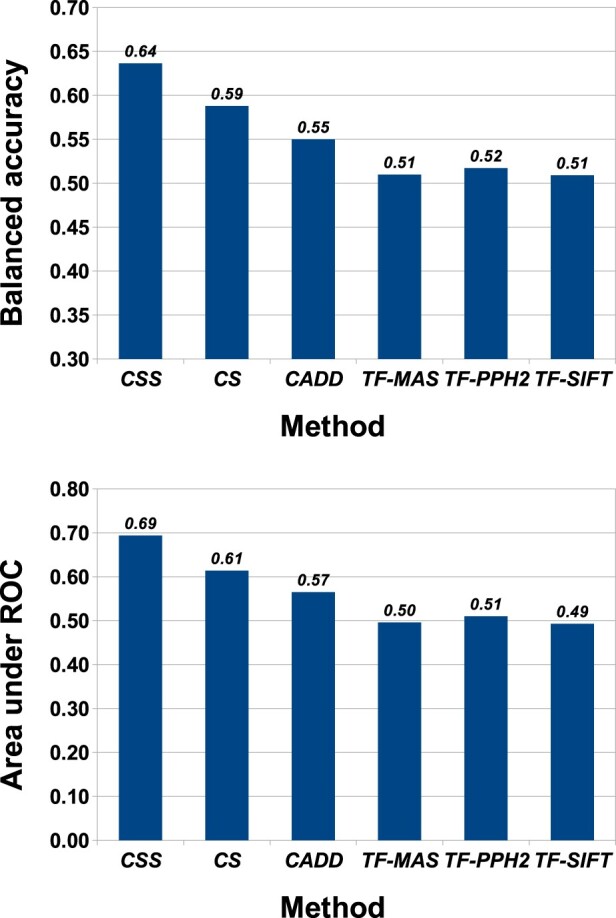
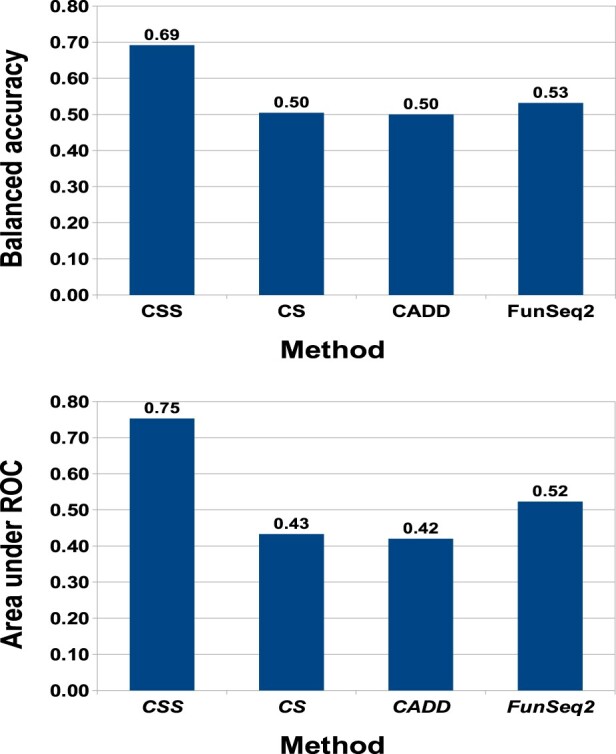
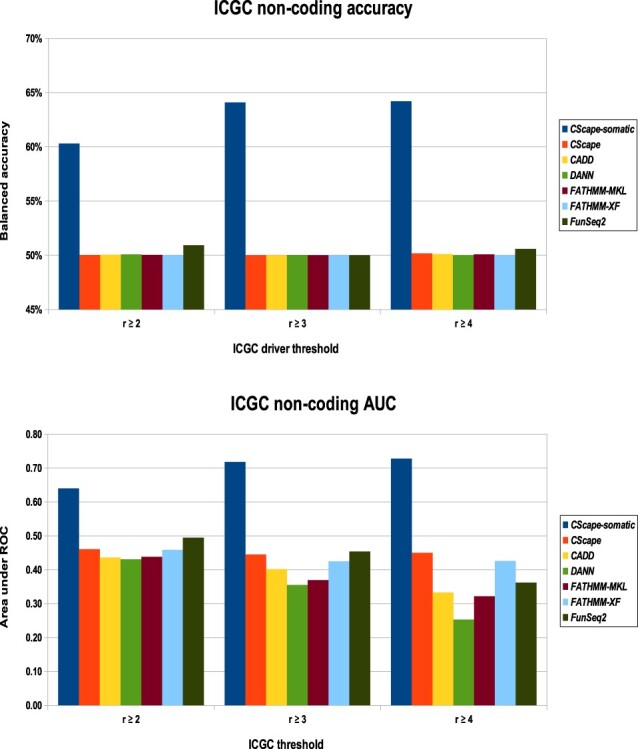
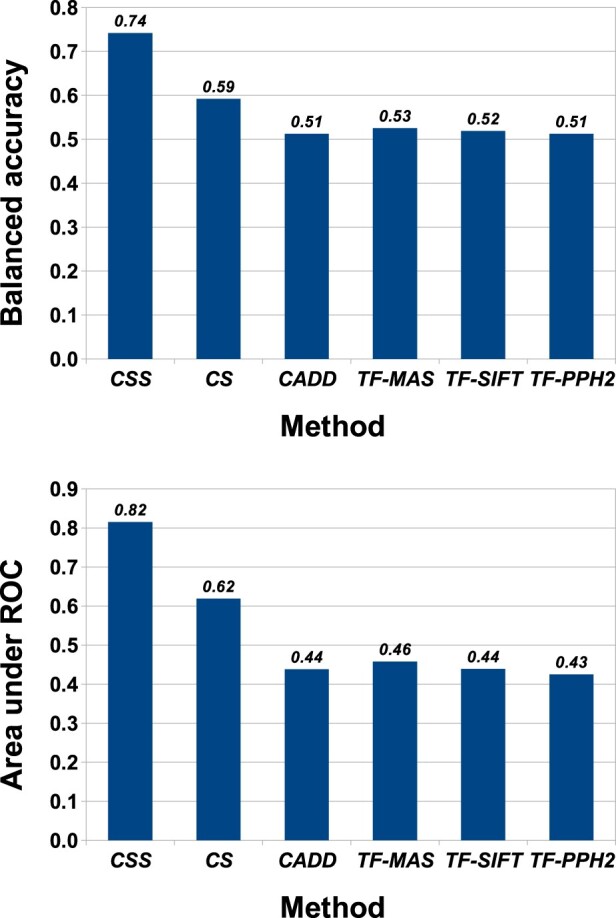
For somatic point mutations in coding and non-coding regions of the genome, we propose CScape-somatic, an integrative classifier for predictively discriminating between recurrent and rare variants in the human cancer genome. In this study, we use purely cancer genome data and investigate the distinction between minimal occurrence and significantly recurrent somatic single point mutations in the human cancer genome. We show that this type of predictive distinction can give novel insight, and may deliver more meaningful prediction in both coding and non-coding regions of the cancer genome. Tested on somatic mutations, CScape-somatic outperforms alternative methods, reaching 74% balanced accuracy in coding regions and 69% in non-coding regions, whereas even higher accuracy may be achieved using thresholds to isolate high-confidence predictions.
Predictions and software are available at http://CScape-somatic.biocompute.org.uk/.
mark.f.rogers.phd@gmail.com or C.Campbell@bristol.ac.uk.
Supplementary data are available at Bioinformatics online.
下一代测序技术加速了人类基因组中单核苷酸变异的发现,刺激了开发预测工具的发展,以分类哪些变异可能在疾病中具有功能,哪些是中性的。最近,我们提出了 CScape 方法,用于区分癌症驱动突变和假定良性变体。对于中性类,该方法依赖于 1000 基因组计划数据库中发现的良性种系变体。因此,区分可以受到种系与体细胞的区分,而不是中性与疾病驱动的区分的影响。这促使我们在本文中考虑仅基于癌症数据在复发和罕见体细胞单点突变之间进行预测性区分,以及这两个体细胞类和种系单点突变之间的区分。
对于基因组编码和非编码区域中的体细胞点突变,我们提出了 CScape-somatic,这是一种用于预测区分人类癌症基因组中复发和罕见变体的综合分类器。在这项研究中,我们仅使用纯粹的癌症基因组数据,并研究人类癌症基因组中最小发生和显著复发的体细胞单点突变之间的区别。我们表明,这种类型的预测性区别可以提供新的见解,并可能在癌症基因组的编码和非编码区域中提供更有意义的预测。在体细胞突变上进行测试时,CScape-somatic 的表现优于替代方法,在编码区域中达到 74%的平衡准确性,在非编码区域中达到 69%,而使用阈值隔离高置信度预测甚至可以获得更高的准确性。
预测和软件可在 http://CScape-somatic.biocompute.org.uk/ 获得。
mark.f.rogers.phd@gmail.com 或 C.Campbell@bristol.ac.uk。
补充数据可在 Bioinformatics 在线获得。