Peacock Andrew J, Ling Yi, Johnson Martin K, Kiely David G, Condliffe Robin, Elliot Charlie A, Gibbs J Simon R, Howard Luke S, Pepke-Zaba Joanna, Sheares Karen K K, Corris Paul A, Fisher Andrew J, Lordan James L, Gaine Sean, Coghlan J Gerry, Wort S John, Gatzoulis Michael A
Scottish Pulmonary Vascular Unit, Golden Jubilee National Hospital, Glasgow, UK.
Sheffield Pulmonary Vascular Disease Unit, Royal Hallamshire Hospital, Sheffield, UK.
Pulm Circ. 2020 Mar 30;10(1):2045894020914851. doi: 10.1177/2045894020914851. eCollection 2020 Jan-Mar.
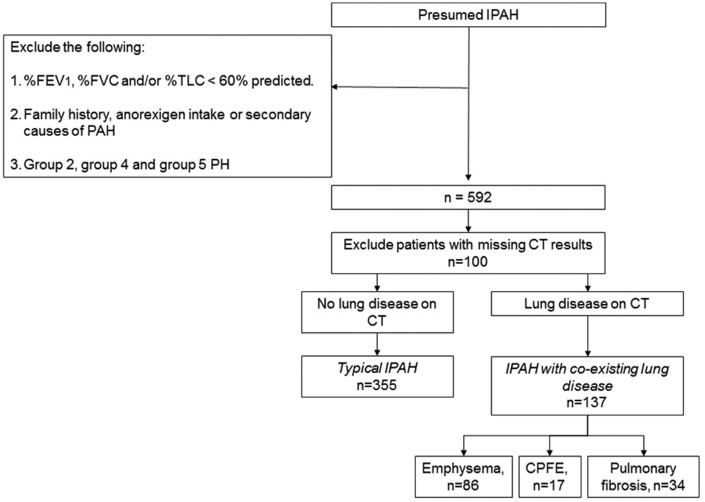
Patients classified as idiopathic pulmonary arterial hypertension (defined as Group 1 on European Respiratory Society (ERS)/European Cardiac Society (ESC) criteria) may have evidence of minor co-existing lung disease on thoracic computed tomography. We hypothesised that these idiopathic pulmonary arterial hypertension patients ( ) are a separate subgroup of idiopathic pulmonary arterial hypertension with different phenotype and outcome compared with idiopathic pulmonary arterial hypertension patients without co-existing lung disease ( ). Patients with ' ' have been eligible for all clinical trials of Group 1 patients because they have normal clinical examination and normal spirometry but we wondered whether they responded to treatment and had similar survival to patients with ' '. We described the outcome of the cohort of patients with ' ' in a previous paper. Here, we have compared incident ' ' patients with ' ' patients diagnosed concurrently in all eight Pulmonary Hypertension centres in the UK and Ireland between 2001-2009. Compared with ' ' ( = 355), ' ' patients ( = 137) were older, less obese, predominantly male, more likely to be current/ex-smokers and had lower six-minute walk distance, lower % predicted diffusion capacity for carbon monoxide, lower mean pulmonary arterial pressure and lower pulmonary vascular resistance index. After three months of pulmonary hypertension-targeted treatment, six-minute walk distance improved equally in ' ' and ' '. However, survival of ' ' was lower than ' ' (one year survival: 72% compared with 93%). This survival was significantly worse in ' ' even after adjusting for age, gender, smoking history, comorbidities and haemodynamics. ' ' patients had similar short-term improvement in six-minute walk distance with anti-pulmonary arterial hypertension therapy but worse survival compared with ' ' patients. This suggests that ' ' are a separate phenotype and should not be lumped with ' ' in clinical trials of Group 1 pulmonary arterial hypertension.
被归类为特发性肺动脉高压(根据欧洲呼吸学会(ERS)/欧洲心脏病学会(ESC)标准定义为第1组)的患者在胸部计算机断层扫描上可能有轻微并存肺部疾病的证据。我们假设这些特发性肺动脉高压患者( )是特发性肺动脉高压的一个单独亚组,与无并存肺部疾病的特发性肺动脉高压患者( )相比,具有不同的表型和预后。“ ”患者符合所有第1组患者的临床试验条件,因为他们临床检查正常且肺功能正常,但我们想知道他们对治疗的反应以及生存情况是否与“ ”患者相似。我们在之前的一篇论文中描述了“ ”患者队列的结局。在此,我们比较了2001年至2009年期间在英国和爱尔兰所有八个肺动脉高压中心同时诊断出的新发“ ”患者与“ ”患者。与“ ”(n = 355)相比,“ ”患者(n = 137)年龄更大,肥胖程度更低,以男性为主,更可能是当前吸烟者/既往吸烟者,且6分钟步行距离更短,一氧化碳预测弥散能力百分比更低,平均肺动脉压更低,肺血管阻力指数更低。经过三个月的肺动脉高压靶向治疗后,“ ”和“ ”患者的6分钟步行距离均有同等程度的改善。然而,“ ”患者的生存率低于“ ”患者(一年生存率:72% 对比93%)。即使在调整年龄、性别、吸烟史、合并症和血流动力学因素后,“ ”患者的生存率仍显著更差。“ ”患者在接受抗肺动脉高压治疗后6分钟步行距离有相似的短期改善,但与“ ”患者相比生存率更差。这表明“ ”是一种单独的表型,在第1组肺动脉高压的临床试验中不应与“ ”归为一类。