Integrated Genomics Laboratory, Oregon Health & Science University, Portland, Oregon, USA.
Department of Molecular & Medical Genetics, Oregon Health & Science University, Portland, Oregon, USA.
Sci Rep. 2020 Apr 14;10(1):6271. doi: 10.1038/s41598-020-62801-6.
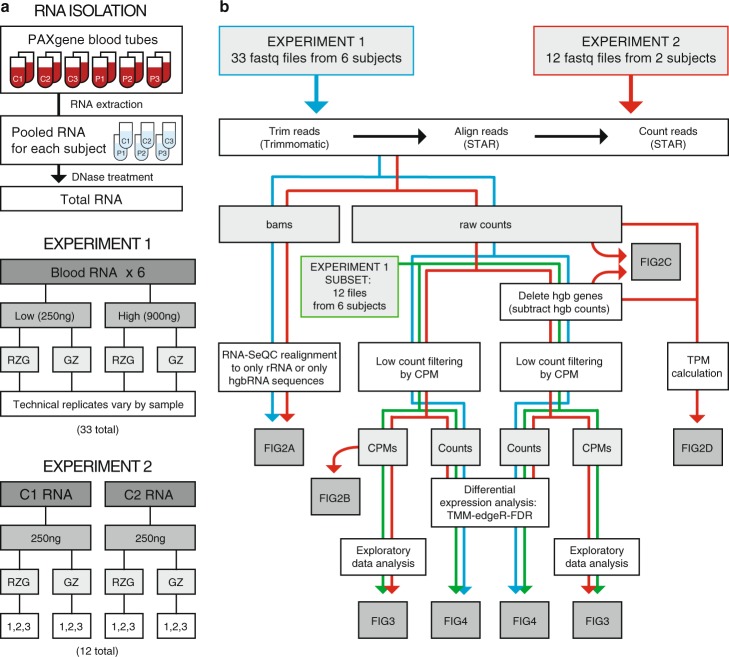
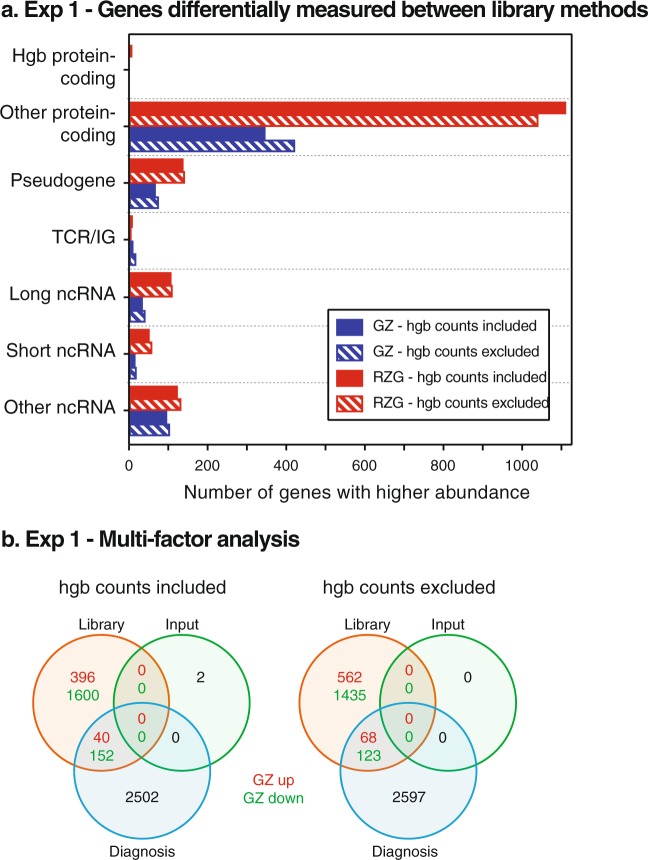
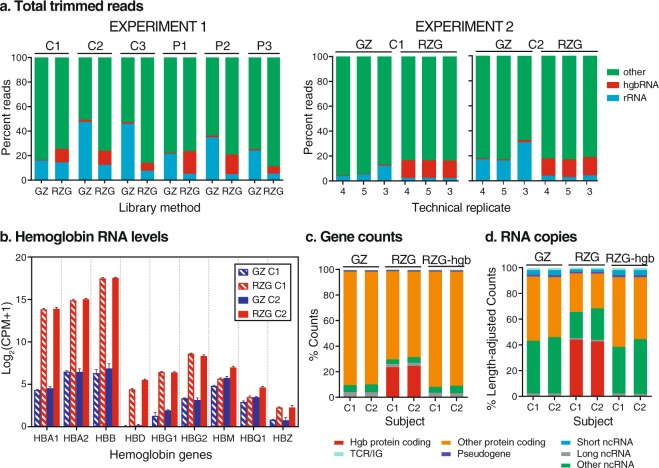
Peripheral blood is a highly accessible biofluid providing a rich source of information about human physiology and health status. However, for studies of the blood transcriptome with RNA sequencing (RNA-Seq) techniques, high levels of hemoglobin mRNAs (hgbRNA) present in blood can occupy valuable sequencing space, impacting detection and quantification of non-hgbRNAs. In this study, we evaluated two methods for preparing ribosomal RNA (rRNA)-depleted sequencing libraries for RNA-Seq of whole blood, one of which is also designed to deplete hgbRNAs. Two experiments were performed: one evaluating library performance across 6 human blood samples and the other examining library reproducibility and performance in a two-subject subset. We find that addition of hgbRNA depletion to the rRNA-depletion protocol for library preparation from blood RNA effectively reduces highly abundant hgbRNA reads; however, it does not result in a statistically significant increase in differentially expressed genes in our patient-control study. Bioinformatic removal of globin gene counts in non-hgbRNA depleted libraries provides improvement in overall performance of these libraries. We conclude that use of a standard ribosomal RNA depletion method for library preparation coupled with bioinformatic removal of globin gene counts is sufficient for reproducible and sensitive measurement of both coding and noncoding RNAs in the blood transcriptome.
外周血是一种高度可及的生物液体,提供了有关人体生理学和健康状况的丰富信息。然而,对于使用 RNA 测序 (RNA-Seq) 技术研究血液转录组学,血液中存在高水平的血红蛋白 mRNA (hgbRNA) 会占用宝贵的测序空间,影响非 hgbRNA 的检测和定量。在这项研究中,我们评估了两种用于制备核糖体 RNA (rRNA) depleted 测序文库的方法,用于全血的 RNA-Seq,其中一种方法还设计用于去除 hgbRNA。进行了两项实验:一项评估了 6 个人类血液样本的文库性能,另一项则在两个受试者亚组中检查了文库的重现性和性能。我们发现,在血液 RNA 的 rRNA depleted 文库制备中添加 hgbRNA 去除,可有效降低高度丰富的 hgbRNA 读段;然而,在我们的患者对照研究中,它并没有导致差异表达基因的数量显著增加。在非 hgbRNA depleted 文库中,通过生物信息学去除珠蛋白基因计数,可提高这些文库的整体性能。我们得出结论,使用标准的核糖体 RNA 去除方法进行文库制备,并结合生物信息学去除珠蛋白基因计数,足以在血液转录组中可重复且灵敏地测量编码和非编码 RNA。