Infectious Diseases and Immune Defence Division, The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC, 3052, Australia.
Department of Medical Biology, The University of Melbourne, Parkville, VIC, Australia.
Sci Rep. 2023 Feb 1;13(1):1859. doi: 10.1038/s41598-023-28218-7.
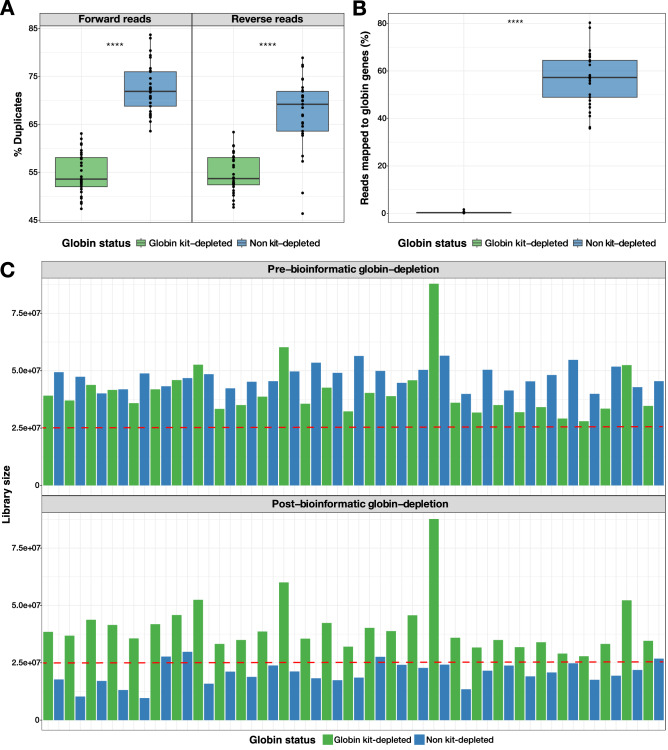
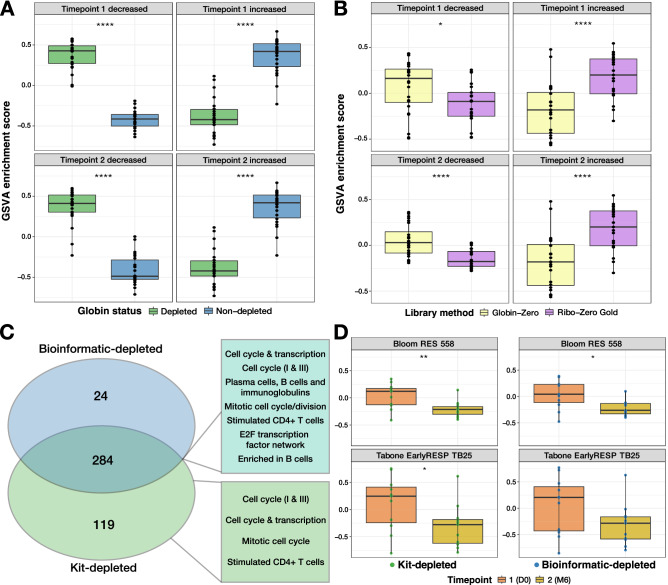
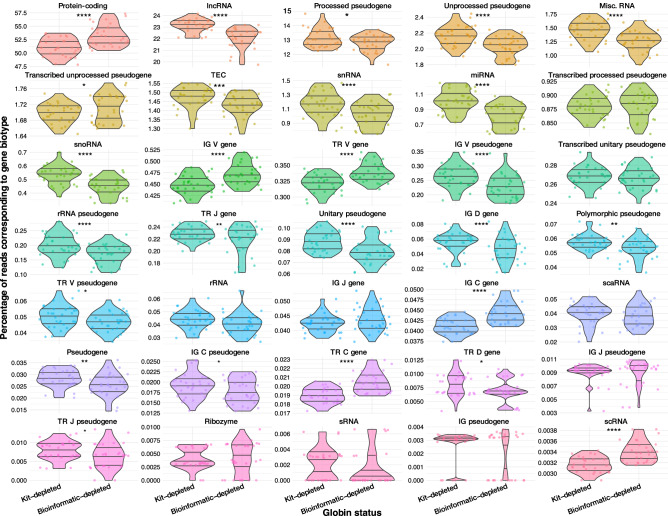
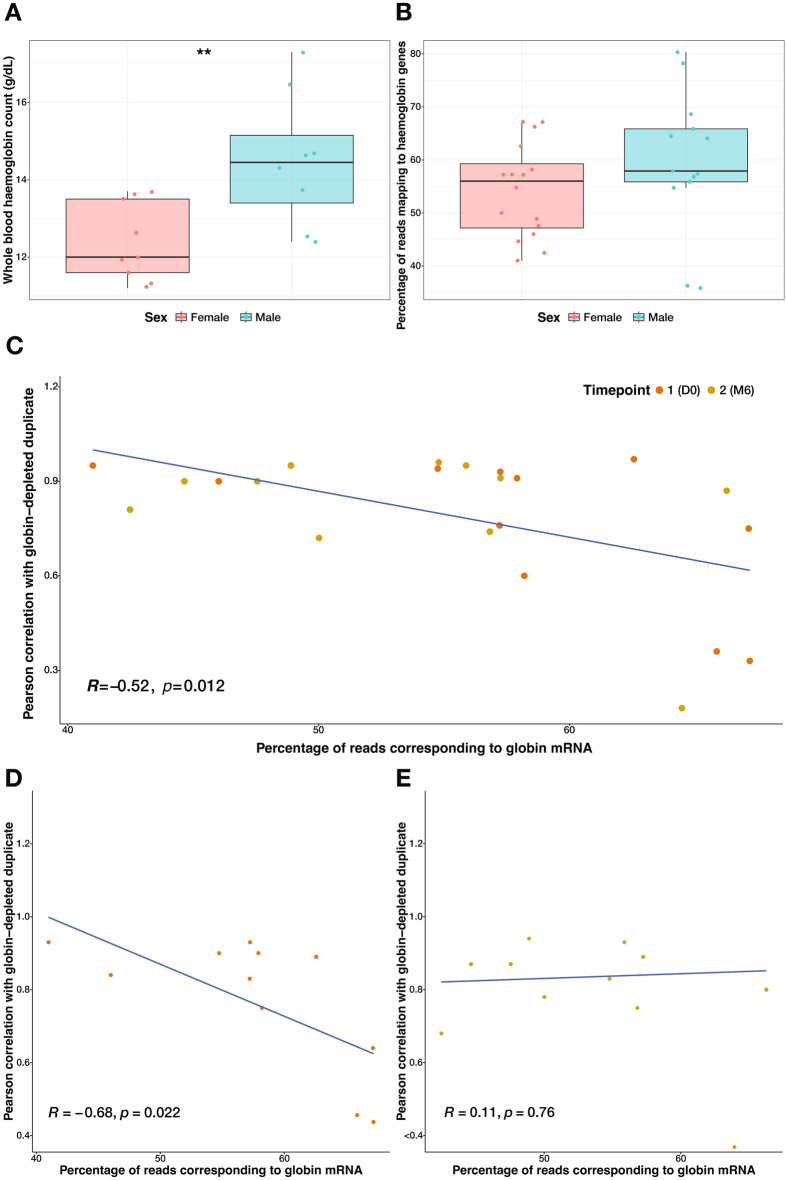
When profiling blood samples by RNA-sequencing (RNA-seq), RNA from haemoglobin (Hgb) can account for up to 70% of the transcriptome. Due to considerations of sequencing depth and power to detect biological variation, Hgb RNA is typically depleted prior to sequencing by hybridisation-based methods; an alternative approach is to deplete reads arising from Hgb RNA bioinformatically. In the present study, we compared the impact of these two approaches on the outcome of differential gene expression analysis performed using RNA-seq data from 58 human tuberculosis (TB) patient or contact whole blood samples-29 globin kit-depleted and 29 matched non-depleted-a subset of which were taken at TB diagnosis and at six months post-TB treatment from the same patient. Bioinformatic depletion of Hgb genes from the non-depleted samples (bioinformatic-depleted) substantially reduced library sizes (median = 57.24%) and fewer long non-coding, micro, small nuclear and small nucleolar RNAs were captured in these libraries. Profiling published TB gene signatures across all samples revealed inferior correlation between kit-depleted and bioinformatic-depleted pairs when the proportion of reads mapping to Hgb genes was higher in the non-depleted sample, particularly at the TB diagnosis time point. A set of putative "globin-fingerprint" genes were identified by directly comparing kit-depleted and bioinformatic-depleted samples at each timepoint. Two TB treatment response signatures were also shown to have decreased differential performance when comparing samples at TB diagnosis to six months post-TB treatment when profiled on the bioinformatic-depleted samples compared with their kit-depleted counterparts. These results demonstrate that failure to deplete Hgb RNA prior to sequencing has a negative impact on the sensitivity to detect disease-relevant gene expression changes even when bioinformatic removal is performed.
当通过 RNA 测序(RNA-seq)对血液样本进行分析时,血红蛋白(Hgb)的 RNA 可占转录组的 70%。由于测序深度和检测生物变异的能力的考虑,血红蛋白 RNA 通常通过基于杂交的方法在测序前被耗尽;另一种方法是通过生物信息学方法从 Hgb RNA 中耗尽读取。在本研究中,我们比较了这两种方法对从 58 例人类结核病(TB)患者或接触者全血样本(29 个珠蛋白试剂盒耗尽和 29 个匹配的非耗尽样本)中获得的 RNA-seq 数据进行差异基因表达分析的结果,其中一些样本是在 TB 诊断时和从同一患者在 TB 治疗后 6 个月采集的。从非耗尽样本中通过生物信息学方法从 Hgb 基因中耗尽(生物信息学耗尽),显著降低了文库大小(中位数=57.24%),并且在这些文库中捕获的长非编码、微、小核和小核仁 RNA 更少。在所有样本中分析已发表的 TB 基因特征,发现在非耗尽样本中未耗尽的 Hgb 基因的读数比例较高时,试剂盒耗尽和生物信息学耗尽对的相关性较差,尤其是在 TB 诊断时。通过直接比较每个时间点的试剂盒耗尽和生物信息学耗尽样本,确定了一组假定的“珠蛋白指纹”基因。当在生物信息学耗尽的样本上比较 TB 诊断时和 TB 治疗后 6 个月的样本时,两个 TB 治疗反应特征也显示出当与试剂盒耗尽样本相比时,其差异性能降低。这些结果表明,即使进行生物信息学去除,在测序前不耗尽 Hgb RNA 也会对检测与疾病相关的基因表达变化的敏感性产生负面影响。