Environmental and Occupational Health Sciences Institute, School of Public Health, Rutgers-the State University of New Jersey, 170 Frelinghuysen Road, Piscataway, NJ, USA 08854, NJ, USA.
Department of Marine and Coastal Sciences, Rutgers-the State University of New Jersey, 71 Dudley Road, New Brunswick, NJ USA 08901.
Gigascience. 2020 Apr 1;9(4). doi: 10.1093/gigascience/giaa038.
Changes to human respiratory tract microbiome may contribute significantly to the progression of respiratory diseases. However, there are few studies examining the relative abundance of microbial communities at the species level along the human respiratory tract.
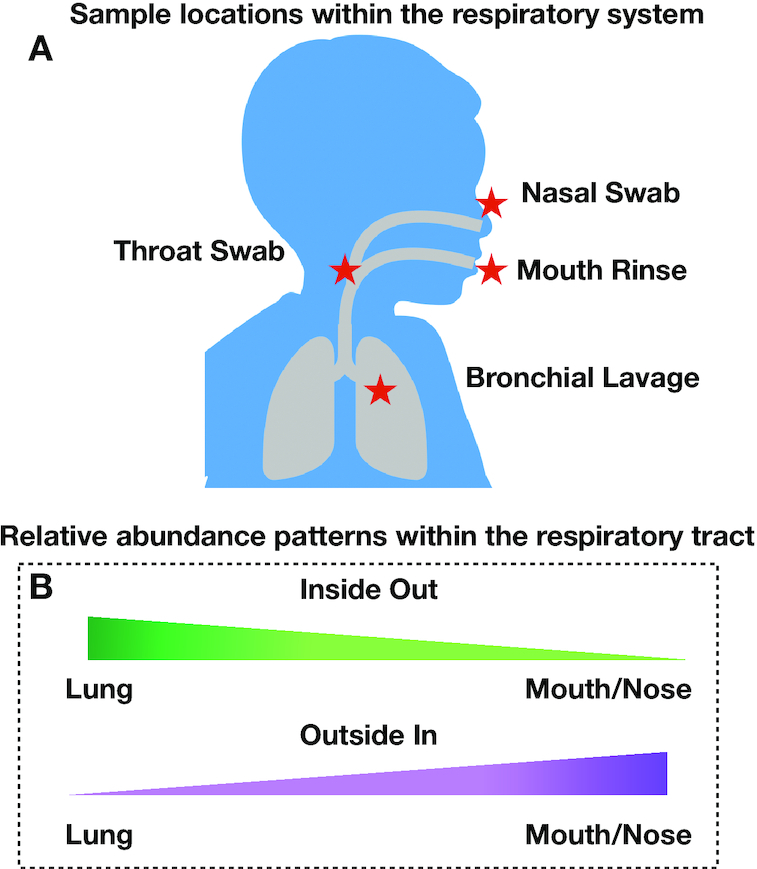
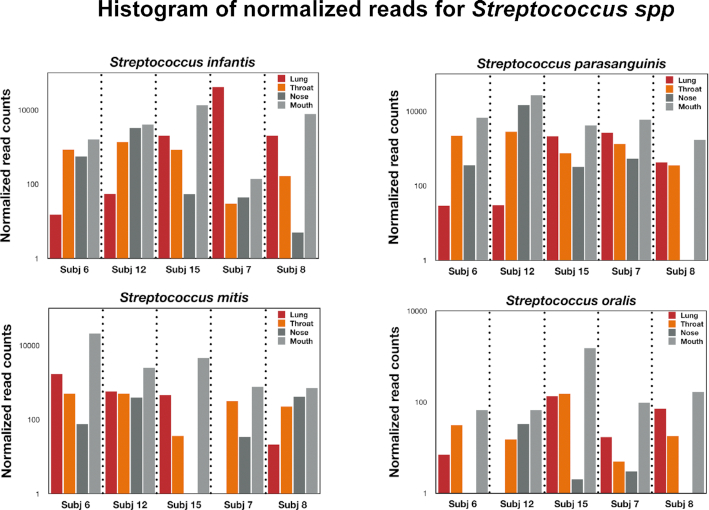
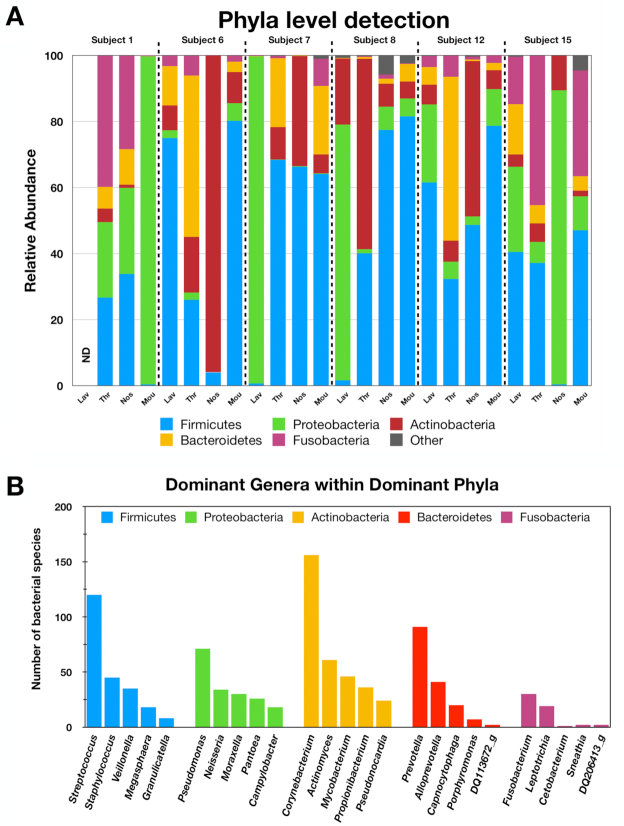
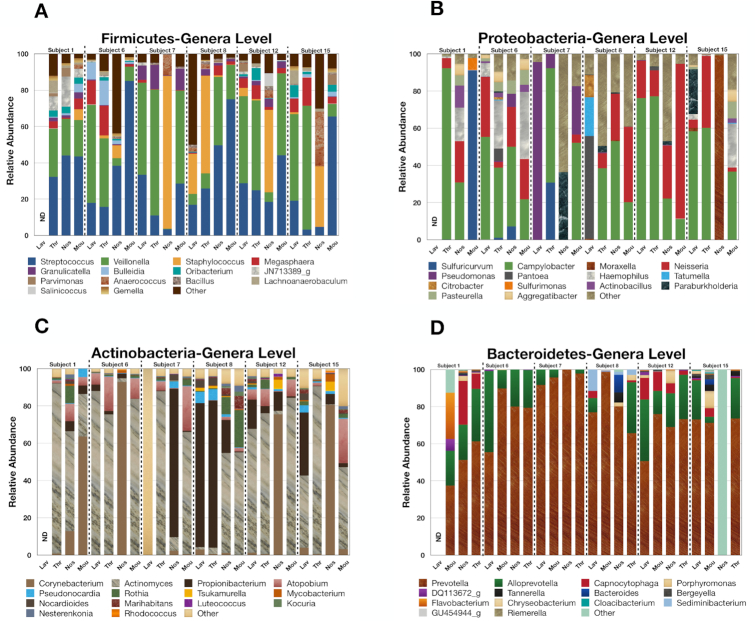
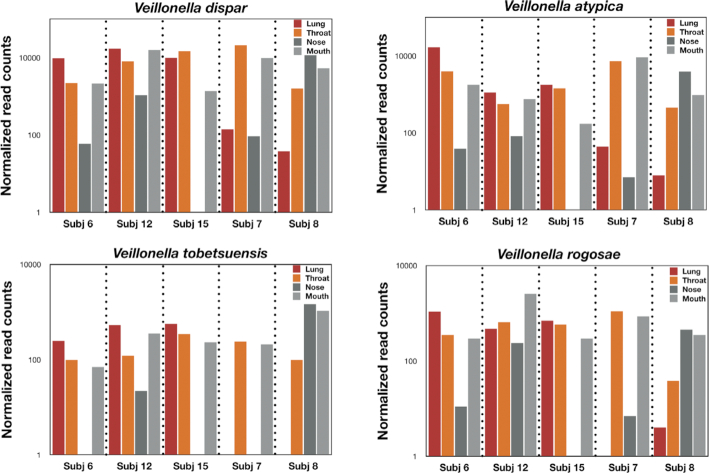
Bronchoalveolar lavage, throat swab, mouth rinse, and nasal swab samples were collected from 5 participants. Bacterial ribosomal operons were sequenced using the Oxford Nanopore MinION to determine the relative abundance of bacterial species in 4 compartments along the respiratory tract. More than 1.8 million raw operon reads were obtained from the participants with ∼600,000 rRNA reads passing quality assurance/quality control (70-95% identify; >1,200 bp alignment) by Discontiguous MegaBLAST against the EZ BioCloud 16S rRNA gene database. Nearly 3,600 bacterial species were detected overall (>750 bacterial species within the 5 dominant phyla: Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes, and Fusobacteria. The relative abundance of bacterial species along the respiratory tract indicated that most microbes (95%) were being passively transported from outside into the lung. However, a small percentage (<5%) of bacterial species were at higher abundance within the lavage samples. The most abundant lung-enriched bacterial species were Veillonella dispar and Veillonella atypica while the most abundant mouth-associated bacterial species were Streptococcus infantis and Streptococcus mitis.
Most bacteria detected in lower respiratory samples do not seem to colonize the lung. However, >100 bacterial species were found to be enriched in bronchoalveolar lavage samples (compared to mouth/nose) and may play a substantial role in lung health.
人类呼吸道微生物组的变化可能对呼吸道疾病的进展有重要影响。然而,目前很少有研究从物种水平上研究呼吸道各部位微生物群落的相对丰度。
从 5 名参与者中采集了支气管肺泡灌洗液、咽喉拭子、口腔冲洗液和鼻腔拭子样本。使用 Oxford Nanopore MinION 对细菌核糖体操纵子进行测序,以确定呼吸道 4 个部位细菌物种的相对丰度。通过 Discontiguous MegaBLAST 对 EZ BioCloud 16S rRNA 基因数据库进行非连续比对,从参与者中获得了超过 180 万条原始操纵子读数,其中约 60 万条 rRNA 读数通过质量保证/质量控制(70-95%鉴定;>1200 bp 比对)。总共检测到近 3600 种细菌物种(>750 种细菌物种存在于 5 个主要门:Firmicutes、Proteobacteria、Actinobacteria、Bacteroidetes 和 Fusobacteria 中。呼吸道各部位细菌物种的相对丰度表明,大多数微生物(95%)是被动从外部输送到肺部的。然而,有一小部分(<5%)细菌物种在灌洗液样本中丰度较高。肺中最丰富的细菌物种是韦荣球菌属和韦荣球菌属,而口腔相关的最丰富的细菌物种是婴儿链球菌和米氏链球菌。
在下呼吸道样本中检测到的大多数细菌似乎并不定植于肺部。然而,在支气管肺泡灌洗液样本(与口腔/鼻腔相比)中发现有>100 种细菌物种被富集,它们可能在肺部健康中发挥重要作用。